|
Paneles de Discussión
Paneais de Discussio Comunicaciones libres
Comunicaçoes livres |
Enfermedad de Alport: un diagnóstico difícil.
Suzanne Meleg-Smith, MD
Investigaciones recientes han demostrado el defecto básico responsable de la Enfermedad de Alport (EA): mutaciones en los genes del colágeno tipo IV. Al mismo tiempo, como el diagnóstico sigue representando un desafío frecuente tanto para el nefrólogo como para el patólogo, analizaré y trataré de aclarar algunas de las dificultades clínicas y patológicas que se presentan en esta enfermedad. Los temas a tratar son:
Definición.
La EA es causada por un defecto congénito del colágeno tipo IV, afectando la membrana basal en diversas localizaciones, especialmente en el glomérulo renal, resultando en presentaciones clínicas variables.
Presentación clínica de la enfermedad de Alport (EA).
Se estima que la incidencia de la EA es de 1:5,000-1:10,000, causando del 1% al 2% de todos los casos de insuficiencia renal terminal.
La manifestación clínica mas importante del la EA, hematuria con cilindros de hematíes, a menudo se inicia en la infancia y frequentemente se asocia a historia familiar positiva para enfermedad renal 1. La hematuria tiende a ser microscópica, pero con episodios de orina sanguinolenta. La proteinuria aparece en general algunos ańos despues y puede acompanarse de síndrome nefrótico.
Cuando los signos y síntomas del paciente se limitan al rińón, el diagnóstico clínico es EA. La designación de síndrome de Alport se emplea cuando, ademas de enfermedad renal, el paciente padece de sordera neurosensorial (revisado en detalle en 2) o de anomalias oculares a nivel de la retina, lente del cristalino o córnea.
En la edad adulta, en los pacientes del sexo masculino, a menudo se observa evolución a insuficiencia renal terminal. En cambio, en las pacientes del sexo femenino, frecuentemente los síntomas son menos severos y la evolución mas benigna.
En ausencia de una historia familiar de enfermedad renal, o de manifestaciones del síndrome a nivel de varios sistemas, el diagnostico clínico de EA es imposible y el nefrólogo recurre a la biopsia renal. Esta permite el diagnostico diferencial entre EA y otras entidades, especialmente la nefropatía IgA, la glomerulopatía que clínicamente, por su manifestación de hematuria, mas se parece a la EA.
Membrana basal glomerular (MBG) normal: colágeno tipo IV.
Aunque existen 6 cadenas del colágeno tipo IV, denominadas de α1 a α6, solo 5 -- de α1 a α5 -- se encuentran en la MBG. Los principales componentes de la MBG son las cadenas α3, α4 y α5, las cuales forman una red supraestructural, donde cada monómero es una triple hélice de 3 cadenas α y se acompaña de proteínas como laminina, proteoglicán y entactin/nidogen. Las cadenas α1 y α2 del colágeno tipo IV están presentes en escasa cantidad en la MBG normal, pero representan uno de los principales componentes del mesangio glomerular y de la cápsula de Bowman. Conviene anotar que la cadena α6 del colágeno tipo IV esta ausente de la MBG, encontrándose solamente en la cápsula de Bowman 3. Los genes que codifican las 6 cadenas del colágeno tipo IV se localizan sobre 3 cromosomas diferentes3, a saber:
Aspectos genéticos de la EA.
Durante las ultimas décadas se ha demostrado que la EA es la consecuencia de mutaciones en los genes del colágeno tipo IV. Estas mutaciones ocasionan cambios en el fenotipo y en la función de la membrana basal, no solamente en el rińón sino también en otros sistemas (vide supra).
La EA es genéticamente heterogénea: la transmisión de la mutación del colágeno tipo IV puede estar ligada al cromosoma X 4 o ser autosómica, recesiva o dominante 5. Y, como no es raro encontrar una biopsia renal con cambios diagnósticos de EA en un paciente sin historia familiar, debemos suponer la ocurrencia de mutaciones genéticas "de novo".
Herencia ligada al cromosoma X. En muchos pacientes con EA con este tipo de herencia, se ha demostrado una mutación del gene COL4A5, el cual codifica la cadena α5 del colágeno tipo IV y esta localizado a nivel de Xq22 6, 7, 8. Se estima que este tipo de herencia se encuentra en el 90 % de las familias con EA9. En casos de herencia ligada al cromosoma X, la severidad de la presentación clínica de la enfermedad en los varones es consecuencia de su estado "hemizigoto" para el cromosoma X. En las mujeres, en cambio, la presentación clínica es variable, siendo asintomáticas cuando el cromosoma X inactivado al azar es el que lleva la mutación del gene COL4A5. En cambio, si el cromosoma X inactivado es el normal, presentaran EA sintomática 10
Se ha reportado gran heterogenicidad alélica entre los pacientes con EA con herencia ligada al cromosoma X, encontrándose mas de 200 mutaciones diferentes del gene COL4A5, incluyendo deleción, inversión, inserción, duplicación, etc.… 11. Aunque recientemente se han descrito algunas familias con una mutación genética idéntica 12, es mas frecuente observar una mutación propia para cada familia.
Jais et al 13 demostraron por primera vez una correlación entre el genotipo y el fenotipo. En resultados obtenidos con la investigación de 195 familias en múltiples países, los autores describieron como mutaciones a nivel del "reading frame" del gen COL4A5 se acompańaban de un 90% de probabilidad de evolución clínica a falla renal terminal antes de los 30 anos de edad.
Herencia autosómica. En un número menos frecuente de familias con EA, se encuentra herencia autosómica, resultado de mutaciones de los genes COL4A3 y COL4A4 --sobre el cromosoma 2 14, los cuales codifican las cadenas α
3 y α4 del colágeno tipo IV15. De gran interés es el descubrimiento de que en varias familias con mutaciones a nivel de los genes COL4A3 o COL4A4, la presentación clínica de los individuos no es idéntica. Las personas homozigóticas para la mutación presentan EA; en cambio, las personas heterozigóticas tienen una presentación menos severa de "hematuria benigna"14, 16.
Los aspectos genéticos de la EA se encuentran discutidos en espańol, con lujo de detalles, en la reciente publicación de Tazon et al17. A pesar de adelantos en nuestros conocimientos de ciencia básica y la disponibilidad de métodos para un diagnostico genético, la mutación no es demostrable en cada paciente y el nefrólogo necesariamente recurre a la biopsia renal.
La biopsia renal en la EA.
A la microscopía de luz, la EA presenta solamente cambios inespecíficos de grado variable en glomérulos e intersticio renal, incluyendo esclerosis glomerular, fibrosis intersticial y presencia de células espumosas. La inmunohistoquímica demuestra que en la EA los depósitos inmunes están ausentes -- en oposición a lo que ocurre en las biopsias de la Nefropatía IgA.
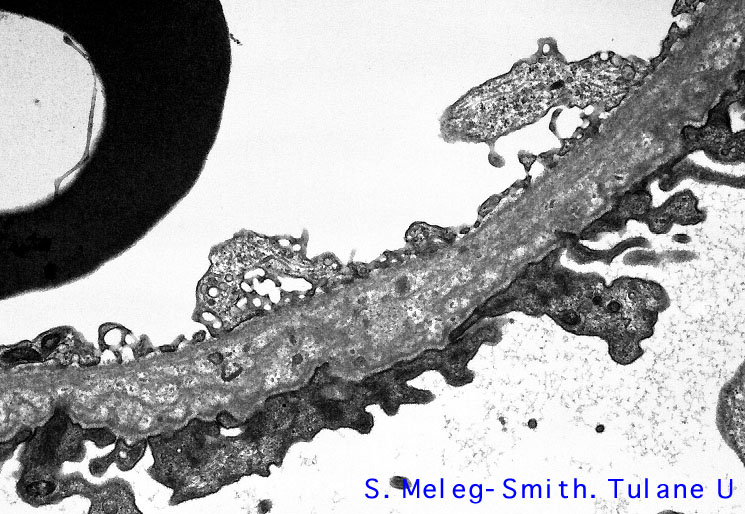
El diagnostico de la EA se hace a base del cambio estructural característico a nivel de la MBG, observado por el microscopio electrónico. El cambio diagnostico de la EA18 es un engrosamiento severo - hasta 3 veces el espesor normal - de la MBG, que adquiere un aspecto de "encaje", como se demuestra en la Microscopía Electrónica 1a.
El aspecto de "encaje" es el resultado de la perdida de la homogeneidad normal de la lamina densa, la cual se reduplica, interrumpe y lamela, tal como se ilustra en la Microscopía Electrónica 2a.
Las áreas con aspecto de "encaje" contrastan con grados variables de adelgazamiento de la MBG - ver Microscopía Electrónica 3 y 4.
Cambios ultraestructurales inespecíficos encontrados en la EA incluyen aumento de la matriz mesangial y ensanchamiento o borramiento de los podocitos. Depósitos densos de tipo inmune están ausentes.
Por lo tanto, el patólogo llega a un diagnostico conclusivo de EA cuando observa extensos cambios de lamelación y engrosamiento de la lámina densa, resultando en un aspecto de "encaje" de la MBG, como se ilustra en la Microscopía Electrónica 5.
Pero, es bien sabido que los cambios patognomónicos no son del mismo grado ni se encuentran en todos los pacientes con EA19, originando dificultades en el diagnostico patológico cuando los cambios de "encaje" son focales y/o escasos y predomina el adelgazamiento de la MBG.
El adelgazamiento de la MBG - ver Microscopía Electrónica 6a. -
Un método sencillo para determinar el espesor de la MBG10 consiste en medir el espesor en 3 puntos equidistantes de la circunferencia de un capilar y calcular el promedio. El capilar puede clasificarse como delgado si el espesor es menor de la medida aceptada para la edad del paciente23, 24. El diagnostico de MBG delgada implica que el cambio es difuso, es decir, se observa en mas de la mitad de los capilares.
El defecto de la MBG en la EA ha sido estudiado con métodos inmunohistoquímicos por medio de antisueros comerciales que identifican las cadenas α3, α4 y α5 del colágeno tipo IV3. Con este método, en un rińón normal, la presencia de cada cadena del colágeno tipo IV origina un patrón lineal continuo en la MBG. En varones con EA con herencia ligada al cromosoma X, la misma prueba inmunohistoquímica muestra la ausencia de la cadena α5 del colágeno tipo IV25. En mujeres con EA ligada al cromosoma X, el patrón inmunohistoquímico obtenido con antisuero a la cadena α5 es diferente tanto del aspecto del rińón normal como del observado en varones con EA: en estas pacientes, se encuentra un aspecto linear discontinuo, interrumpido10, 26.
Sorpresivamente, en algunos pacientes con mutación del gene COL4A5, en quienes la transcripción de las cadenas α
3 y α4 del colágeno tipo IV es normal27, la inmunohistoquímica no confirma la presencia de cadenas α
3 y α4 28. Asimismo, el estudio inmunohistoquímico de la biopsia de pacientes con EA con herencia autosómica puede no demostrar la cadena α
5 29. Para comprender estos hallazgos, hay que recurrir a los trabajos de Gunwar et al 30, quienes en estudios de la estructura supramolecular del colágeno tipo IV, encontraron que la unión covalente normal entre las cadenas α3, α4 y α5 se interrumpe aun cuando la mutación afecta solamente una de las cadenas; el resultado es un ensamblaje defectuoso de las cadenas, con falta de visualización inmunohistoquímica de las 3. Por lo tanto, los métodos inmunohistoquímicos no permiten diagnosticar la mutación exacta responsable de la EA. Es interesante recordar que al faltar las cadenas mas importantes del colágeno tipo IV --α3, α4 y α5 -- la MBG en la EA esta constituida por cadenas α1 y α227. Esta MBG funciona adecuadamente en la nińez, pues las cadenas α
1 y α2 sustituyen por las otras cadenas faltantes. Pero, eventualmente la lamina densa anormal da origen a las lamelaciones que resultan en el aspecto de "encaje" de la MBG observada en edad adulta31.
Correlación clínico-patológica en la EA.
El patólogo hace fácilmente un diagnostico de EA cuando el microscopio electrónico demuestra, en forma difusa, un aspecto de "encaje" de la MBG32, ilustrado en la Microscopía Electrónica 8.
En estos casos, el fenotipo de la MBG implica un pronostico reservado, con evolución posible a insuficiencia renal terminal. Puede representar EA con herencia ligada al cromosoma X o herencia autosómica con estado homozigotico.
En cambio, la MBG extensivamente adelgazada, como se observa en la Microscopía Electrónica 9, representa un verdadero reto para el patólogo32
En familias con EA con herencia ligada al cromosoma X, una biopsia renal con adelgazamiento difuso de la MBG es altamente sugestiva del diagnostico mientras que el pronostico no es predecible. En mujeres cuya biopsia muestra MBG adelgazada, el pronostico tiende a ser benigno33. En cambio, en hombres se ha visto que el mismo cambio no es garantía de buen pronostico: una biopsia con el único cambio de adelgazamiento de la MBG en la nińez puede evolucionar a lamelacion en una biopsia en edad mayor y acompańarse de enfermedad renal terminal34.
Finalmente, en un hombre adulto, el cambio ultraestructural de adelgazamiento de la MBG es sugestivo de EA de herencia autosómica, con estado heterozigotico para mutaciones del gen COL4A3 o del COL4A4, e implica un buen pronostico. Posiblemente estos casos corresponden a la entidad clínica anteriormente denominada "hematuria familiar benigna"35.
En conclusión, espero haber demostrado que mientras una presentación clínica típica sugiere EA, la confirmación requiere la identificación de la mutación genética. La biopsia renal, además de contribuir a la determinación del pronóstico, seguirá con su papel importante en el diagnostico hasta el momento en que las pruebas genéticas se conviertan en estudios rutinarios, con capacidad de demostrar la mutación genética del colágeno tipo IV en cada uno de los pacientes. Bibliografía
1.- Pirson, Y., Making the diagnosis of Alport's syndrome. Kidney Int, 1999. 56: p. 760-0775.
2.- Kashtan, C.E. and A.F. Michael, Alport Syndrome. Kidney Int, 1996. 50: p. 1445-1463.
3.- Kashtan, C., Alport syndrome and thin glomerular basement membrane disease. J Am Soc Nephrol, 1998. 9: p. 1736-1750.
4.- Lemmink, H., et al., The clinical spectrum of type IV collagen mutations. Hum Mutat, 1997. 9: p. 477-99.
5.- Toren, A., et al., Genetic linkage of autosomal-dominant Alport syndrome with leukocyte inclusions and macrothrombocytopenia (Fechtner syndrome) to chromosome 22Q11-13. Am J Hum Genet, 1999. 65: p. 1711-7.
6.- Tryggvason, K., et al., Molecular genetics of Alport syndrome. Kidney Int, 1993. 43: p. 38-44.
7.- Martin, P., et al., High mutation detection rate in the COL4A5 collagen gene in suspected Alport syndrome using PCR and direct DNA sequencing. J Am Soc Nephrol, 1998. 9: p. 2291-2301.
8.- Zhou, J., et al., Complete amino acid sequence of the human a5(IV) collagen chain and identification of a single-base mutation in exon 23 converting glycine 521 in the collagenous domain to cysteine in an Alport syndrome patient. J Biol Chem, 1992. 267(18): p. 12475-12481.
9.- Mazzucco, G., et al., Ultrastructural and immunohistochemical findings in Alport's syndrome: A study of 108 patients from 97 Italian families with particular emphasis on COL4A5 gene mutation. J Am Soc Nephrol, 1998. 9: p. 1023-1031.
10.- Meleg-Smith, S., et al., X-linked Alport syndrome in females. Hum Pathol, 1998. 29: p. 404.
11.- Antignac, C., et al., Deletions in the COL4A5 collagen gene in X-linked Alport syndrome. Characterization of the pathological transcripts in nonrenal cells and correlation with disease expression. J Clin Invest., 1994. 93(3): p. 1195-1207.
12.- Barker, D., et al., A mutation causing Alport syndrome with tardive hearing loss is common in the western United States. Am J Hum Genet, 1996. 58: p. 1157-1165.
13.- Jais, J., et al., X-linked Alport syndrome: Natural history in 195 families and genotype-phenotype correlations in males. J Am Soc Nephrol, 2000. 11: p. 649-657.
14.- Heidet, L., et al., Structure of the Human Type IV Collagen Gene COL4A3 and Mutations in Autosomal Alport Syndrome. J Am Soc Nephrol, 2001. 12(1): p. 97-106.
15.- Mochizuki, T., et al., Identification of mutations in the A3(IV) and A4(IV) collagen genes in autosomal recessive Alport syndrome. Nature Genetics, 1994. 8: p. 77-81.
16.- Lemmink, H., et al., Benign familial hematuria due to mutation of the type IV collagen alpha4 gene. J Clin Invest, 1996. 98: p. 1114-8.
17.- Tazon, B., E. Ars, and R. Torra, [The Alport syndrome]. Nefrologia, 2003. 23 Suppl 1: p. 29-39.
18.- Hinglais, N., J. Grunfeld, and E. Bois, Characteristic ultrastructural lesion of the glomerular basement membrane in progressive hereditary nephritis (Alport syndrome). Lab Invest, 1972. 27: p. 473-487.
19.- Rumpelt, H.-J., Hereditary nephropathy (Alport syndrome): correlation of clinical data with glomerular basement membrane alterations. Clin Nephrol, 1980. 13(5): p. 203-207.
20.- Buzza, M., D. Wilson, and J. Savige, Segregation of hematuria in thin basement membrane disease with haplotypes at the loci for Alport syndrome. Kidney Int, 2001. 59(5): p. 1670-1676.
21.- Dische, F., Measurement of glomerular basement membrane thickness and its application to the diagnosis of thin-membrane nephropathy. Arch Pathol Lab Med, 1992. 116: p. 43-49.
22.- Basta-Jovanovic, G., et al., Morphometric analysis of glomerular basement membranes (GBM) in thin basement membrane disease (TBMD). Clin Nephrol, 1990. 33: p. 110-114.
23.- Steffes, M., et al., Quantitative glomerular morphology of the normal human kidney. Lab Invest, 1983. 49: p. 82-86.
24.- Vogler, C., A. McAdams, and S. Homan, Glomerular basement membrane and lamina densa in infants and children: An Ultrastructural evaluation. Pediatr Pathol, 1987. 7: p. 527-534.
25.- Kleppel, M.M., et al., Immunochemical studies of the Alport antigen. Kidney Int, 1992. 41: p. 1629-1637.
26.- Hara, M., et al., Immunohistochemical Localization of Glomerular Basement Membrane Antigens in Various Renal Diseases. Virchows Arch, 1986. 408(4): p. 403-419.
27.- Heidet, L., et al., Glomerular expression of type IV collagen chains in normal and X-linked Alport syndrome kidneys. Am J Pathol, 2000. 156: p. 19001-1910.
28.- Nakanishi, K., et al., Expression of Type IV Collagen a3 and a4 Chain mRNA in X-Linked Alport Syndrome. J Am Soc Nephrol, 1996. 7: p. 938-945.
29.- Gubler, M.C., et al., Autosomal recessive Alport syndrome: immunohistochemical study of type IV collagen chain distribution. Kidney Int, 1995. 47(4): p. 1142-1147.
30.- Gunwar, S., et al., Glomerular basement membrane. J Biol Chem, 1998. 273: p. 8767-8775.
31.- Miner, J.H., Of laminins and delamination in Alport syndrome. Kidney Int, 2003. 63: p. 1158-1159.
32.- Meleg-Smith, S., Alport Disease: a review of the diagnostic difficulties. Ultra pathol, 2001. 25: p. 193-200.
33.- Moghal, N., et al., Coexistence of thin membrane and Alport nephropathies in families with haematuria. Pediatr Nephrol, 1999. 13: p. 778-781.
34.- Cangiotti, A., et al., Evolution of glomerular basement membrane lesions in a male patient with Alport syndrome: Ultrastructural and morphometric study. Nephrol Dial Transplant, 1996. 11: p. 1829-1834.
35.- Abe, S., et al., Thin basement membrane syndrome in adults. J Clin Pathol, 1987. 40: p. 318-322.
|