Key words: review, renin molecule, renin gene, renin biosynthe-sis, juxtaglo-merular apparatus, calcium, cyclic nucleotides, osmotic forces, baro-receptor, macula densa, renal nerves, angiotensin II, prostaglandins, endothelial factors.
INTRODUCTION
Tigerstedt and Bergman (1898) showed, that extracts of rabbit kidney cortex contained a vasopressor substance, which they named renin. It was subsequently established that renin is an enzyme, which acts upon a substrate of high molecular weight -angiotensinogen - that is produced in large quantities by the liver. Renin cleaves the decapeptide angiotensin I (angI) from the substrate. The physiologically active prod-uct, angiotensin II (angII) (Braun-Menendez et al. 1939), emerges after removal of the two end-terminal amino acids by an angio-tensin-converting enzyme (ACE) which is located mainly to the surface of endo-thelial cells in all vascular beds (Caldwell et al. 1976) and which is responsible for conversion of angI to angII in the plasma. In tissues there is evidence for non-ACE-dependent conversion (mainly by chymase) of angI to angII in humans and several other species, but not in rats and rabbits (Hollenberg 2000). The effects of angII are initiated after binding of the peptide to specific receptors, of which two subtypes have been identified (AT-1 and AT-2) (Whitebread et al. 1989, Inagami et al. 1999). AngII affects blood pressure in a number of ways: the total vascular resistance is increased by direct effects on the resistance vessels, and by facilitation of sympathetic nerve transmission. Salt and water excretion is influenced by stimulation of the adrenal production of aldosterone, by effects on renal proximal tubules, glomerular arterioles, and renal medullary blood flow (Seldin & Giebisch 1992.). In addition, angII has a stimulatory effect on the thirst centre and ADH secretion in the CNS (Phillips & Sumners 1998). The renin-angiotensin system (RAS) has received attention as a local growth factor that may be involved in the pathogenesis of hypertrophy of heart (Wollert & Drexler 1999) and blood vessels during chronic heart failure and hypertension. The RAS thus plays an important role for the control of blood pressure and extracellular fluid homeostasis.
In spite of the existence of local renin-systems in various tissues (e.g. reproduction organs, adrenal glands, CNS), the concentration of active renin in plasma is determined mainly by the rate of renin secretion from the kidneys, which are the only organs that contain substantial amounts of readily releasable active renin.
THE RENIN MOLECULE AND GENE
Human renal renin is a single-chained, glycosylated carboxy peptidase that belongs to the aspartyl proteinase family, which also includes pepsin, chymosin and lysosomal cathepsins. By contrast to these enzymes renin is very substrate-specific and is active at neutral pH. The active site of the renin molecule is located in a cleft between two nearly symmetrical lobes. Two aspartic acid residues at the active site are essential for the catalytic function. Human renin has a molecular weight of about 41.000, an isoelectric point of 5.2-5.8, and a pH optimum for the reaction with angiotensinogen of 5.5-6.0 (Taugner & Hackenthal, 1989, Morris1992)
The human genome contains one renin gene, which is located to chromosome number 1. It is about 12 kbases in size and consists of ten exons and nine introns (Imai et al. 1983). In analogy with other structural genes there are several putative promoter regions in the 5'-flanking region from where the RNA-polymerase may initiate transcription. In the human kidney transcription begins at one site only and it results in one type of pre-pro-mRNA. The transcriptional rate of the renin gene is probably stimulated directly by cyclic AMP (after nerve stimulation; infusion of isoprenaline), which influences enhancer sequences of the renin gene (Baxter et al. 1991, Morris et al. 1994, Ying et al. 1997). Transcription is inhibited by angII: treatment with ACE-inhibitors stimulates transcription in a way that is reversed by angII (Johns, 1990). A low-sodium diet stimulates transcription by a mechanism that may or may not (Morgan et al. 1991) involve the macula densa. Also, a protein-rich diet is associated with an increase in rat renal renin mRNA (Rosenberg et al. 1990).
A substantial fraction of the regulation of renin synthesis takes place at the level of mRNA. Thus, using primary cultures of mouse JG-cells, Chen et al. (1993) reported that the increased renin mRNA levels induced by cAMP are due in part to a selective increase in renin mRNA stability. Sinn and Sigmund (1999) studied the human carcinoma-derived cell line Calu-6, and found a similar significant effect of cAMP on human renin-mRNA stability. Except from these reports, the knowledge of mRNA metabolism in JG-cells is largely unknown.
RENIN BIOSYNTHESIS.
Translation of renin mRNA gives preprorenin, which consists of 406 amino acids (Imai et al. 1983). During transfer to the endoplasmic reticulum the signal peptide (20 amino acids) is cleaved off, and the molecule is glycosylated (Baxter et al. 1991). The glycosylation affects the biological halftime of the molecule (Kim et al. 1989), but may also slow down the processing of prorenin to renin (Imai et al. 1983). The pro-part of the molecule forms a plug in the cleft between the two lobes of the molecule and hinders the admission of renin-substrate to the catalytic site. After transfer to the Golgi apparatus, prorenin is packed into protogranules that pinches off the transmost Golgi cisterns. A part of the protogranules is not modified any further and proceeds to be secreted as prorenin via the constitutive pathway (Pratt1987), while others coalesce to form larger secretory granules. As mentioned, the renin molecule is related to lysosomal enzymes, and the renin-containing secretory granules have several characteristics in common with lysosomes (Taugner et al. 1985). They have an acid pH; they contain typical lysosomal enzymes such as cathepsin B and D, acid phosphatase and ß-glucuronidase. These enzymes, most probably cathepsin B (Wang et al. 1991), assist in removal of the prosegment from the renin molecule thereby exposing the active site. Consequently, the amount of active renin increases while the granules mature whereas the amount of prorenin decreases. The secretory mechanism has been the subject of several studies.
Thus, renin release from rat afferent arterioles in vitro occurs as a quantal event with an amount of renin discharged per episode, which corresponds to the calculated values for the renin contents of single juxtaglomerular cell granules (about 1 million renin molecules per granule) (Skøtt, 1986). Punctiform sites of fusion between the cell and granule membrane are observed after in vivo stimulation of secretion (Taugner et al. 1984). Exocytosis leads to addition of membrane material to the surface of the cell. This has been measured by whole-cell patch clamp on single JG-cells as an increase in cell membrane capacitance (Friis et al. 1999). In addition, after acute stimulation of the renin system, there are fewer renin granules in the afferent arterioles than in controls (Rasch et al. 1998). These observations strongly suggest that renin storage granules are secreted by exocytosis. The secretory events occur with the same probability to the luminal and abluminal side of the cells.
The mature renin granules constitute a store of active renin, which is secreted in a regulated fashion. The rate of prorenin release by the constitutive pathway seems to depend on the rate of renin synthesis. In human plasma all of the active renin comes from the kidney. After "activation" of blood plasma (treatment with e.g. trypsin), the renin activity is increased by a factor of 10. A large proportion of this inactive renin is prorenin. After nephrectomy the active renin disappears from the blood, while the concentration of inactive renin activity is halved. In rats similar findings have been reported, but here it has been shown that the "inactive renin" remaining after nephrectomy is neither prorenin nor renin (Kim et al. 1991), suggesting that all prorenin also comes from the kidney.
The physiological significance of plasma prorenin has not been clarified. It has been suggested that the circulating inactive renin is a pool from which tissue specific enzymes could recruit active renin with local effect. Although several in vitro procedures have proved capable of activating renin (cold, acidic pH and proteolytic digestion), the experimental evidence for such activation in vivo is still lacking (Reudelhuber et al. 1998).
MORPHOLOGY
The majority of renal renin is synthesized, stored, and released by cells located in the afferent arteriole normally close to the entry into the renal corpuscle. The cells containing renin (juxtaglomerular (JG)/epithelioid/renin-positive cells) are developed by metaplastic transformation of smooth muscle cells in the lamina media of the arteriolar wall. The JG-cells have well-developed secretory machinery: large nucleus, distinct endoplasmic reticulum and Golgi complex, and a number of large mature secretory granules, a few protogranules and many small clear vesicles. Macular gap-junctions couple the JG-cells to each other, to neighbouring Goormaghtigh cells, and to smooth muscle cells further upstream the afferent arteriole (Taugner & Hackenthal, 1989).
There are about 5-20 JG-cells in each afferent arteriole, but the number varies considerably and depends on several factors such as the location in the kidney (higher renin content in cortical arterioles), the age (in fetal life the renin positive segment extends as far as the arcuate arteries but after partus the arterial tree quickly attains adult renin profile (Gomez et al 1989)), the salt balance (low-sodium diet increases the renin positive segment of the afferent arteriole), and certain pathophysiological conditions (Taugner & Hackenthal, 1989). Finally, various medications, such as ACE-inhibition, AT-1 blockade and heavy diuretic treatment, are associated with excessive renin production, and extension of JG-cells upstream towards the cortical radiate artery. The plasticity of the renin-producing segment of the afferent arteriole thus enables the RAS to adapt to the differing demands put on the secretory capacity. The cellular mechanism responsible for the transformation is not known.
The afferent arteriole forms a part of the juxtaglomerular apparatus. This complex is formed by a) the two glomerular arterioles, b) a specialized plaque of tubular epithelial cells called the macula densa (Zimmermann 1933), which is located in the wall of the thick ascending limb of Henle at the point where the tubule returns to its parent glomerulus, and c) modified interstitial cells (Goormaghtigh cells/extraglomerular mesangium) which fill out the space between the glomerular tuft and the macula densa (Goormaghtigh 1932).
INTRACELLULAR REGULATION OF RENIN SECRETION
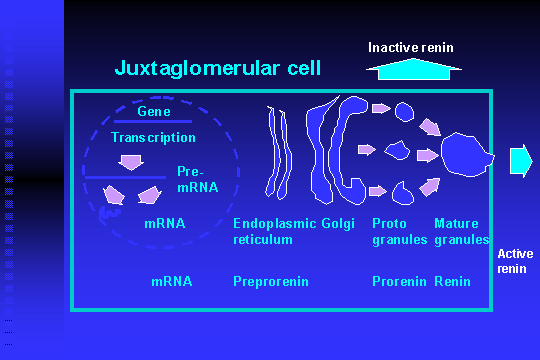
Figure 1. Pathway of renin synthesis in a juxtaglomerular cell
The most well-known intracellular messenger molecules involved in the control of renin secretion are calcium and the cyclic nucleotides, cAMP and cGMP. Calcium plays a role as a negative second messenger, which mediates the inhibition of renin release that results from exposure to vasoconstrictors such as angiotensin II, endothelin, ADH, or a1 adrenergic agonists. Cyclic AMP is a stimulatory second messenger, which mediates the increase in renin release that results from stimulation of the cells with beta-1 adrenergic agonists, prostaglandin I2 and prostaglandin E2, dopamine, and adrenomedullin. Cyclic GMP may stimulate or inhibit renin release depending on circumstances, and is the second messenger of nitric oxide and natriuretic peptides such as atrial natriuretic peptide.
Calcium in juxtaglomerular cells.
The calcium concentration (Ca2+) in the cytosol of the resting JG-cell is 50-100 nM, and it can be increased transiently by mobilization of calcium from intracellular stores, and by opening of calcium channels in the cell membrane. Receptor activation (e.g. by angII) may stimulate both mechanisms (Kurtz & Penner 1989).
Hormone-activated release of calcium from the intracellular stores of JG-cells follows the classical pathway: receptor-activation stimulates phospholipase C (PLC) enzyme activity by means of a presumed regulatory G-protein. Activated PLC then liberates inositol tri-sphosphate (IP3) and diacylglycerol (DAG) from phospholipids located to the inside of the cell membrane. DAG causes stimulation of protein kinase C. IP3 causes release of calcium from intracellular stores. After stimulation by angII this calcium release oscillates (Kurtz & Penner 1989). Depolarisation of the membrane potential reduces the frequency of the calcium oscillations. In this way receptor-agonist interactions and electrical events may act in concert to regulate intracellular calcium. After receptor activation a transmembrane calcium entry pathway is important for the increased level of calcium in the steady state that follows the initial oscillations. The calcium entry pathway has not been clarified. Pharmacological blockade of voltage dependent calcium channels stimulates renin release from a variety of preparations (Churchill, 1985) while electrophysiological measurements on JG-cells have not been able to provide evidence for the existence of such channels (Kurtz et al. 1990).
Calcium and renin secretion
Much experimental evidence speaks in favour of an inhibitory role for calcium in renin release. Renin release from different preparations is inhibited by facilitation of calcium entry and is enhanced by calcium entry antagonists (Hackenthal et al.1990, Churchill, 1985, Baumbach & Leyssac 1977). In addition, calcium-mobilizing hormones such as ADH and angII (Vandongen & Peart, 1974) inhibit renin secretion in a calcium dependent way. Calcium often exerts its physiological effects after binding to specific molecules such as calmodulin, and calmodulin antagonists seem to increase renin secretion (Hackenthal et al.1990).
The JG-cells possess calcium-activated chloride channels (Kurtz & Penner 1989). Because the membrane potential of JG-cells is probably below the equilibrium potential of chloride, activation of these channels (and potassium channels?) may lead to efflux of chloride (and potassium). If potassium follows chloride, water must follow for osmotic reasons and the JG-cells shrink. Because renin release is sensitive to water fluxes (Skøtt 1988), this ability of intracellular calcium to influence JG-cell volume may play a role in the control of renin secretion (Kurtz 1990). If potassium does not follow chloride, the membrane potential will depolarise, and the intracellular chloride concentration may decrease. Renin release from permeabilised JG-cells is sensitive to the intracellular chloride concentration, and thereby variations in the intracellular chloride concentration may be a potential mediator for calcium-induced changes in renin secretion (Jensen & Skøtt, 1994). In accordance with this suggestion, angiotensin II-induced inhibition of renin secretion is counteracted by inhibition of calcium-activated chloride channels (Nabel et al. 1999), and after permeabilization of the cell membrane renin release is no longer stimulated by a decrease in the calcium concentration, while it is highly sensitive to the chloride concentration (Jensen & Skøtt 1994).
Calcium may also interact with a sub-cortical network of myofilaments (Taugner et al. 1988), which hinders access of secretory granules to the cell membrane. In concert with this idea Park et al. (1998) suggested that phosphorylation of MLC20 by Ca2+-calmodulin-dependent MLCK inhibits renin release. Results from isolated glomeruli suggest that the stimulation of renin release caused by low external calcium involves both an anion-sensitive mechanism, and recruitment of secretory granules to the cell membrane (Skøtt & Jensen, 1992). Thus, calcium may affect renin release by several mechanisms.
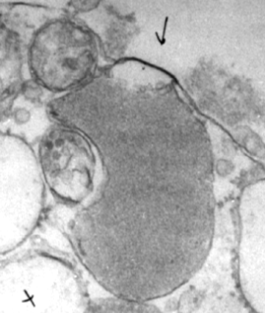
Figure 2. The secretory process in a juxtaglomerular cell
Cyclic AMP
Stimulation of the membrane-bound enzyme adenylyl cyclase followed by generation of cyclic AMP from cytosolic ATP is an important step in the stimulus-response coupling of many hormones. In JG-cells receptor mediated stimulation of adenylyl cyclase (nerve stimulation, ß-agonists) acts to increase the intracellular concentration of cyclic AMP and results in stimulation of renin secretion. Augmentation of renin secretion is also observed when cyclic AMP is increased experimentally, independently of receptor activation. The concentration of cAMP in the cells is determined not only by its formation rate, but also by the rate of degradation. In JG-cells there is evidence that phosphodiesterases of type 3 and type 4 are most significant in this degradation. Thus, inhibition of either phosphodiesterase result in an increase in renin secretion in conscious animals and humans, and in isolated perfused kidneys (Chiu & Reid, 1996, Kurtz et al., 1998). PDE-3 is of special interest because it is under the negative control of cyclic GMP and potentially links the two cyclic nucleotides together. Most of the effect of cAMP on renin secretion is probably mediated by protein kinase A. Cyclic AMP is hardly an obligatory second messenger because renin release can also be stimulated independently of cyclic AMP (Pardy et al. 1989). Cyclic AMP may interact with calcium regulation. Thus, the calcium liberating effect of IP3 is suppressed by the concomitant presence of cyclic AMP in the cytosol (Kurtz & Penner 1989) and cyclic AMP is not able to enhance secretion if calcium is increased experimentally.
Cyclic GMP is generated from cellular GTP by membrane bound or soluble guanylyl cyclase. Cyclic GMP is a potent vasodilator and is produced intracellularly by smooth muscle cells, which have been stimulated by nitric oxide (through the soluble guanylyl cyclase) or atrial natriuretic peptide (ANP) acting on membrane receptors. An increased level of cyclic GMP in renal JG-cells is associated with stimulation of renin release (Hackenthal et al.1990), inhibition of renin secretion (Kurtz et al. 1986), or no change in renin release. Cyclic GMP may stimulate renin secretion by interaction with the cAMP formation through inhibition of PDE-3 as suggested by studies in intact animals, isolated perfused kidneys and by patch-clamp on isolated JG-cells (Chiu & Reid, 1996, Kurtz et al. PNAS, 1998, Friis et al 1999). The pathway that mediates the inhibitory effects of cGMP is less clear. In JG-cells the G-kinase cGKI is located in the cytoplasm, and cGKII is membrane-bound to secretory granules or the cell membrane (Gambaryan et al. 1998). Because cGMP analogues inhibit renin release in JG-cells lacking cGKI, but not cGKII, the latter kinase is a good candidate as a mediator of the inhibitory effect of cGMP on renin release (Wagner et al. 1998).
Osmotic forces
Secretion of renin from JG-cells is highly sensitive to changes in extracellular osmolality (Frederiksen et al. 1975, Skøtt 1988). Thus, exposure of JG-cells in vitro to acute hyposmolality is associated with an increased frequency of renin discharges, and with swelling of secretory granules, adjoinment of secretory granules with cell membrane, and areas of fusion (Skøtt 1986, Skøtt & Taugner 1987). Patch clamp data show that under these conditions the JG-cell capacitance increases, reflecting addition of membrane to the surface after exocytosis of renin granules (Friis et al. 1999). Vesicle swelling induced by uptake of KCl may be involved in the renin exocytic event (Skøtt & Jensen 1989; Park et al. 1991, Skøtt & Jensen 1992, Jensen et al. 1999). In several cell types extensive granular swelling, which results from volume phase transition of the granule matrix, may be necessary for stabilization and dilation of the fusion pore and thereby assist in content extrusion (Zimmerberg et al. 1987) Because extracellular hyposmolality causes increased secretion in JG-cells, water flux must stimulate secretion from JG-cells by a mechanism that precedes the fusion between granule and cell membrane. In JG-cells with permeabilized cell membranes, cell volume changes are avoided, but the osmotic sensitivity is maintained (Jensen & Skøtt 1993). This suggests a role of water fluxes between cytosol and the granules in the late steps of exocytosis.
Ion channels and carriers of the JG-cells
The juxtaglomerular cells share basic electrophysiological properties with most smooth muscle cells. The resting membrane potential of cells in vitro is about 70 mV -80 mV, and is primarily a potassium-diffusion potential. Whole-cell patch-clamp experiments on JG-cells have allowed identification of two different types of voltage dependent potassium channels. One is activated by depolarisation (to -10 mV or more positive potentials) and allows potassium to flow out of the cell (delayed outward rectifier), while the other is activated by hyperpolarization and lets potassium into the cell (anomalous inward rectifier). By such direct experiments it has not been possible to identify voltage dependent channels for calcium or sodium (Kurtz & Penner 1989, Kurtz et al. 1990). Increases in intracellular calcium concentration of the JG-cells lead to opening of chloride channels (Kurtz & Penner 1989), Thus, any stimulus to the cells that causes intracellular calcium to increase (e.g. angII, AVP, alpha-adrenergic stimulation) also causes an increase in chloride conductance, and thereby depolarisation of the cells, and probably also cell shrinkage and/or loss of chloride. The precise significance of these channels in the control of renin secretion has not been determined with certainty.
Indirect experiments where renin secretion was measured after treatment of JG-cell preparations with different chemical agents have suggested the existence in the JG-area of anion exchange mechanisms (HCO3/Cl (Skøtt & Jensen 1989), Na/H exchange mechanism (Kurtz et al. 1991), which probably is activated by protein kinase C, a Na/Ca exchange mechanism (Churchill, 1985), and voltage dependent calcium channels (Churchill, 1985). Blockers of Na/K/2Cl cotransport (loop diuretics) have no direct effect on the JG-cells and their acute stimulatory effect on renin release in intact kidneys is probably mediated by the macula densa (Itoh & Carretero 1985, Lorenz et al. 1991). An immunofluorescence study has revealed the presence of an Na-K-Cl cotransporter of type BSC2 in the juxtaglomerular afferent arteriole (Kaplan et al. 1996).
It may be concluded that the JG-cells as regards electro-physiological properties and ion translocation mechanisms have many similarities to other cells. Activation of some mechanisms may influence renin secretion (e.g. calcium channels), others probably have "house-keeping" functions in the cells, including volume regulation. For most of them the definitive role for control of secretion has not been settled.
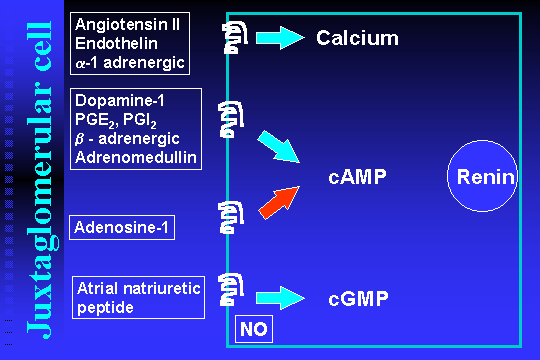
Figure 3. Hormonal control of renin secretion from juxtaglomerular cells
INTRARENAL MECHANISMS OF RENIN SECRETION.
In keeping with the complex homeostatic role of the renin-angiotensin system, renin secretion is under physiologic control by a number of mechanisms. Release is stimulated by decreases in arteriolar pressure, by increases in the activity of the renal nerves, by a signal originating in the tubular fluid at the macula densa, and it is probably also influenced by the local concentrations of a variety of substances including prostaglandins, catecholamines, angII, and adenosine.
The renal baroreceptor mechanism
Reduction of the renal blood perfusion pressure by partial clamping of the renal artery leads to a renin-dependent hypertension, which is probably similar to the pathological conditions observed with renal artery stenosis (Goldblatt et al. 1934, Tobian 1960). In experiments with both anaesthetized and conscious dogs it has been demonstrated that stepwise reductions in renal arterial perfusion pressure lead to an increase in renin release before detectable changes in renal blood flow or glomerular filtration rate (Skinner et al. 1963, Kirchheim et al. 1988). These results suggest the existence of a renal baroreceptor, which stimulates renin release when the renal perfusion pressure drops. Similar results from experiments on dogs with denervated, non-filtering kidneys suggest that the phenomenon is independent of the macula densa mechanism and renal nerve activity (Blaine et al. 1971). The relationship between renin secretion and the perfusion pressure is not linear; secretion increases steeply when the perfusion pressure is decreased below 90 mmHg, i.e. only a little below the mean arterial pressure. Stimulation of the renal nerves causes increased sensitivity of the baroreceptor mechanism: a smaller drop in perfusion pressure is necessary to stimulate renin release when the renal nerve activity is increased or when the postsynaptic receptors are stimulated pharmacologically with alpha-agonists (Ehmke et al. 1989). This nerve-mediated effect on threshold pressure may be involved in the pathogenesis of congestive heart failure, where early sympathetic activation has been suggested to cause renin-mediated volume retention (Kirchheim et al. 1988).
The intrarenal mechanism, which is responsible for the pressure sensitivity is generally assumed to include a vascular receptor located to the wall of the afferent arteriole, which is stimulated by a drop in wall tension. The wall tension depends both on the transmural pressure gradient and the arteriolar diameter. This is in accordance with the observed stimulation of renin secretion when the transmural pressure declines (drop in perfusion pressure or increase in interstitial pressure (ureteral obstruction)) and when the arteriolar diameter is increased without a concomitant decrease in pressure (Fray 1980). The baroreceptor mechanism has been suggested to involve stretch-sensitive cells in the arteriolar wall. Mechanical stretch inhibits basal and forskolin-stimulated renin release and renin mRNA accumulation in cultured rat JG cells and human renin-expressing CaLu-6 cells (Carey et al. 1997) while acute experiments on isolated pressurized afferent arterioles have given conflicting results (Bock et al. 1992, Salomonsson et al. 1991).
Clamping of the renal artery in the intact kidney is accompanied by marked changes in salt excretion even before GFR is changed. The macula densa mechanism may, therefore, contribute to the effects on renin release observed after reduction of the renal perfusion pressure.
The macula densa mechanism
The close anatomical relationship between the macula densa and the renin-containing cells in the afferent arteriole led Goormaghtigh (1937) to propose that the glomerular circulation is automatically regulated by the composition of the urine passing the macula densa segment. Based on whole-kidney studies Vander & Miller (1964) suggested that an increase in the NaCl load at the macula densa would inhibit renin release, and this prediction was supported by the finding of a decreased renin concentration in efferent arteriolar blood or in proximal tubular fluid when the loop perfusion rate was increased by microperfusion (Leyssac 1986). Compelling evidence for this hypothesis came from studies using a technique by which single juxtaglomerular apparatuses were microdissected from rabbits and the tubule segment containing the macula densa was perfused, while simultaneously the entire juxtaglomerular apparatus was superfused and the fluid was collected for renin measurement (Skøtt & Briggs 1987). Results from this preparation have shown that a low NaCl concentration at the macula densa stimulates renin release, and vice versa. The effect is reversible and is independent of osmolality. Luminal application of loop-diuretics mimics the effect of low NaCl, implying that it is the transport rate of NaCl across the apical membrane of the macula densa cells, which determines the renin secretory response. The maximal sensitivity to variations in NaCl-concentration is within the physiological range (25-60 mM). Ion substitution experiments suggest that chloride is more important than sodium for the macula densa response (Lorenz et al. 1991, Skøtt & Briggs 1987, Lorenz et al. 1990). NaCl is taken up by an Na-K-2Cl cotransporter of the BSC1 isoform (Kaplan et al. 1996). It is not yet known how the rate of NaCl transport across the apical membrane of the MD-cells is transmitted to the JG-cells.
The JG-interstitium is poor in capillary and lymphatic drainage, and the macula densa has low water permeability (González et al. 1988). Consequently the composition (osmolality?) of the interstitial fluid may be influenced by the macula densa transport rate of NaCl (Persson et al. 1988). Variations in interstitial ion composition resulting from changes in transport rate may directly influence the JGcells or influence the local generation of prostaglandins (Okuda et al. 1989), or NO from mesangial cells (Tsukuhara et al. 1994).
Two enzymes - cyclooxygenases I and II - are responsible for the production of prostaglandins from arachidonic acid. In the rat there is cortical expression of COX-2 in macula densa and the thick ascending limb of Henle (Harris et al. 1994). In humans expression of COX-2 immunoreactive protein was detected in endothelial and smooth muscle cells of arteries and veins and in podocytes (Komhoff et al. 1997, Khan et al. 1998). These authors did not find COX-2 in the human macula densa while Nantel et al. (1999) found expression in macula densa in elderly humans. The expression of COX-2 in macula densa cells is increased by a low-salt diet (Harris et al. 1994). Conversely, inhibition of prostaglandins with an unspecific cyclooxygenase inhibitor (indomethacin) or COX-2 specific inhibitor, blocks acute macula densa-mediated renin release (Greenberg et al 1993, Traynor et al. 1999). Both acute and chronic stimulation of the macula densa mechanism may therefore include activation of the prostaglandin system.
NO-synthase of the neuronal isoform (nNOS or NOS-I) is also expressed in the macula densa cells (Wilcox et al 1992. Mundel et al. 1992) and the expression of NOS-I is upregulated with a low salt diet. Singh et al. 1996). Production of NO appears to be a critical determinant of the stimulation of renin secretion by low NaCl at the macula densa because the NaCl dependency of renin secretion from the isolated juxtaglomerular apparatus is abolished in the presence of a NOS-blocker in the tubular lumen (He et al. 1995), and NOS-I blockers inhibit the stimulation of renin release by low-salt diet and furosemide treatment (Beierwaltes 1995, 1997).
Local production of adenosine may well play a role in the signalling of the tubuloglomerular feedback mechanism, but has probably only a minor role in macula densa-mediated renin release (Schnermann 1998).
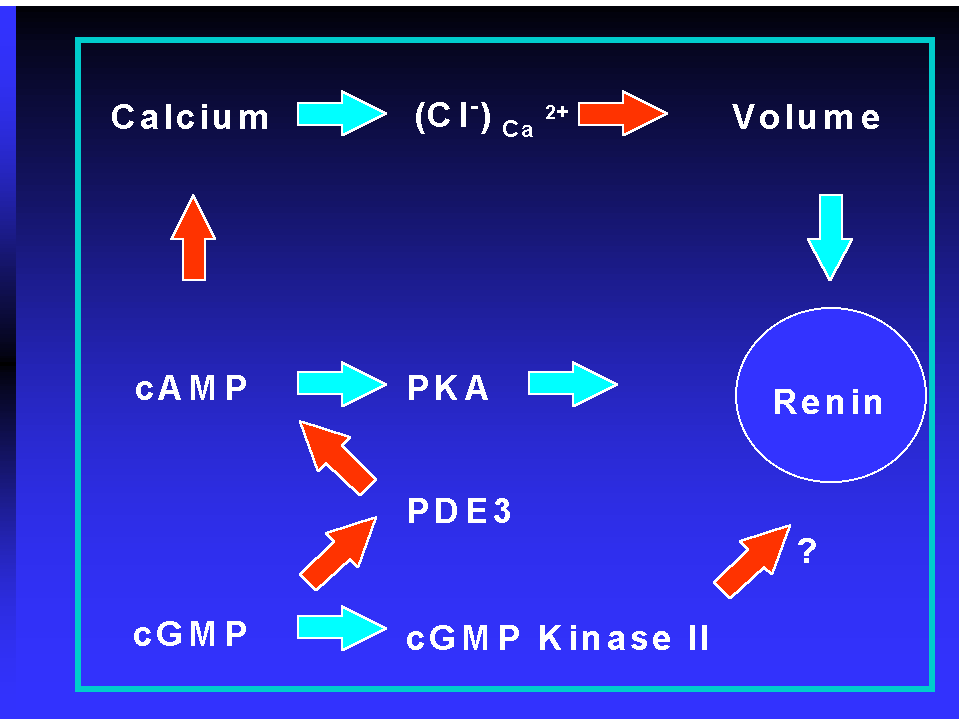
Figure 4 Interaction between intracellular messengers in the control renin of renin secretion
Renal nerves and renin secretion
The renal blood vessels and tubular structures, including the juxtaglomerular apparatus, are innervated by sympathetic nerves, which mainly contain norepinephrine. Activation of the renal nerves has multiple effects: vasoconstriction, reduced GFR, increased proximal tubular fluid reabsorption and enhanced renin secretion (DiBona & Kopp, 1997)
The afferent renal nerves have been stimulated directly by application of depolarising current pulses, and indirectly by compression of the carotid artery, which activates the sinus caroticum and is followed by an elevated sympathetic outflow. Stimulation of the renal nerves in either way leads to an augmented renin secretory activity. The renin secretory response is observed even at low levels of nerve stimulation that have no effects on vascular tonus, tubular function or GFR. The increase in renin secretion is inhibited by ß-adrenergic antagonists or by denervation (Holdaas et al. 1981, DiBona & Kopp, 1997). Consistent with this, renin release from a number of in vitro preparations is stimulated by ß-adrenergic agonists. In isolated cells producing renin the production of cyclic AMP increased in response to ß-agonists. Altogether there is good evidence that ß-receptors are located on the JG-cells and that they operate by activation of adenylyl cyclase There is good evidence that prevailing renal sympathetic nerve activity can modulate the magnitude of the renin secretion rate response to stimulation of the renal vascular baroreceptor and the tubular macula densa receptor mechanisms (DiBona & Kopp, 1997).
The transmitter substance of the sympathetic nerve terminals is norepinephrine, which is known to stimulate both alpha and ß-receptors. Treatment of JG-cells with low concentrations of norepinephrine leads to an enhanced renin release, which can be inhibited by ß-antagonists. Higher concentrations of norepinephrine have been reported to decrease renin secretion from in vitro preparations of JG-cells in a way that can be inhibited by alpha-antagonists. The alpha-receptor mediated decrease in secretion rate is probably mediated by an intracellular increase in Ca++. In the integrated organism the renin secretory response of alpha-receptor stimulation is the opposite, probably because of secondary effects by alpha-stimulation on renal hemodynamics, tubular reabsorption, and the release of locally acting hormones (e.g. prostaglandins) (Keeton & Campbell, 1980).
Other putative transmitters have been discovered in renal nerve endings, and some of these might affect renin release. In other systems some of the substances have been shown to activate adenylyl cyclase (dopamine, VIP, Calcitonin related peptide) while others have been reported to inhibit the adenylyl cyclase (neuropeptide Y). Any physiological role in the control of renin secretion remains to be established (Hackenthal et al.1990).
Endothelial factors
The identification of the endothelial cell layer as a production site of vasoconstrictors (endothelin) and vasodilators (nitric oxide, Endothelial-derived hyperpolarizing factor) has focussed attention on the possible involvement of these factors in the local control of renin secretion.
Endothelins appear to inhibit the renin system acting via Ca2+- and protein kinase C-related mechanisms by an effect directly on the JG-cells. (Rakugi et al. 1988, Takagi et al. 1989, Kurtz et al. 1991, Wagner et al. 1998). Thus, endothelins inhibit cyclic-AMP induced renin gene expression and renin release in cultured mouse juxtaglomerular cells with no difference between endothelin (ET)-1, ET-2, and ET-3 (Ritthaler et al. 1995, Ritthaler et al. 1996). On the other hand, infusion of low-levels of endothelin in man (Vierhapper et al. 1990, Sørensen et al. 1994) or dog (Sandgaard & Bie, 1996) has no effect on plasma renin or angiotensin II levels.
The effect of NO on renin release is mediated through activation of soluble guanylyl cyclase and formation of cGMP. As mentioned above, cGMP may stimulate renin release by inhibition of PDE-3, and subsequent increase in the cAMP concentration (Chiu and Reid, 1996, Kurtz et al. 1998). Inhibition may be mediated through cGKII (Wagner et al. 1998). This dual intracellular pathway may explain the diversity in results obtained with different model systems: perfusate from carotid arteries treated with acetylcholine to stimulate NO-production inhibits renin release from dog kidney slices (Vidal et al. 1988), and blockade of NO formation stimulates renin release from rat renal cortical slices (Beierwalters et al. 1992), and in awake dogs (Salazar et al. 1992). By contrast, blockade of NO formation reduces renin release in anaesthetized rats (Johnson & Freeman, 1992), isolated perfused kidneys (Gardes et al. 1992), and cocultures of endothelial cells and mouse JG-cells (Kurtz et al. 1991).
Humoral factors
A multitude of hormones, cytokines and neurotransmitters affect renin release, but in many cases it has been difficult to define precisely the role of a humoral substance. Different routes of administration in various preparations probably results in varying concentrations of the agents around the JG-cells, and the complex interaction of different control systems may also contribute to conflicting results.
Hormones that interact with adenylyl cyclase-coupled receptors and augment the intracellular concentration of cyclic AMP stimulate renin release. This group of agents comprise ß-agonists, and prostaglandins. Histamine stimulates adenylyl cyclase by interaction with H2-receptors on the JG-cells (Pinet et al. 1987). The physiological role of histamine in renin release is not clear. Hormones, which inhibit adenylyl cyclase generally, inhibit renin secretion. Thus adenosine interacts with A1-receptors on the JG-cells to cause inhibition of renin release (Kurtz et al. 1988, Pfeifer et al. 1995).
The kidney proximal tubules have the ability to decarboxylate circulating L-dopa into dopamine. Activation of this endogenous dopamine production by infusion of the dopamine precursor gamma-L-glutamyl-L-dopa reduces the plasma renin activity in humans. It is unsettled what type of dopamine receptor which mediates this inhibition (MacDonald et al. 1988). On the other hand, infusion of dopamine itself into humans causes a stimulation of renin release, which is mediated by dopamine 1 (DA-1) receptors (Horton et al. 1990). A similar DA-1 mediated stimulation by dopamine has been reported from a number of in vivo and in vitro experiments (Horton et al. 1990, Kurtz, et al. 1988). In rat JG-cells the receptors are of the DA-1A subtype (Yamaguchi et al. 1997) and they have been found, among other places, in the renin storage granules (O'Connell, 1995). D3 and D4 dopamin receptors are also expressed in JG-cells (Sanada et al. 1997) and activation of the D3 receptors inhibit forskolin-stimulated renin release. Disruption of the D3 receptor increases renal renin production and produces renal sodium retention and renin-dependent hypertension (Asico et al. 1998). Thus, most evidence suggests that infused dopamine acts on (DA-1A) receptors on JG-cells to stimulate renin release, while endogenous dopamine produced by the proximal tubules may have the opposite effect, probably by an effect on D3 receptors.
The cellular effect of agents that increase the intra-cellular calcium concentration (angII, endothelin, ADH, alpha-agonists) is inhibition of renin release. AngII interacts with specific receptors and activates calcium channels and initiates generation of IP3, which liberates calcium from intracellular stores. In addition, the transcriptional rate of the renin gene is also suppressed by angII. These direct effects of angII act together to cause a tonic inhibition of renin secretion. This is uncovered when the formation or the effect of angII is inhibited (ACE-inhibitors or AT-1 receptor blockers) and renin secretion and synthesis increase to reach high levels (Kono et al. 1981). These effects of angII on renin synthesis have been thought to be mediated by direct effects on the JG-cells, but Matsusaka et al (1996) developed chimeric mice carrying regional null mutations of the angiotensin type 1A (AT1A) receptor in some areas of the kidney and intact receptors in other areas. The degree of JG hypertrophy/hyperplasia and the expression of renin mRNA and protein turned out to be identical between Agtr1a +/+ and Agtr1a -/- cells, indicating that the local interaction between angiotensin and the AT1 receptor on the JG cells has little functional contribution to the feedback regulation of JG renin synthesis. AngII has an indirect inhibitory effect on renin release by inducing a rise in blood pressure, by the salt-retaining effect of aldosterone and by an effect on baroreceptor sensitivity (Wong et al. 1993). Systemic angII reaches the kidney by the blood stream, but, in addition, a local generation of angII is likely to occur because both renin substrate and ACE are present in the renal interstitium. AngII has even been demonstrated within the secretory granules of JG-cells, but it is not clear whether it has been taken up from the interstitium or whether it has been produced intracellularly.
Antidiuretic hormone does not seem to play any significant physiological role in renin release (Taugner & Hackenthal, 1989, Hackenthal et al.1990) although a direct depolarising effect on JG-cell membrane potential has been observed (Bührle et al. 1985).
Oxytocin infusion stimulates renin secretion in rats (Sjøquist et al. 1999) and it has been suggested that pituitary OT secretion during hypotension or hypovolemia may serve to support blood pressure by enhancing activation of the renin-angiotensin system via a beta-adrenergic receptor-dependent mechanism (Huang et al. 2000).
In the kidney activation of the calcium-sensitive enzyme phospholipase A2 cause liberation of arachidonic acid from the cell membrane, and initiates, thereby, synthesis of prostaglandins, leukotriens and tromboxane. The kidney vasculature produces predominantly prostacyclin (PGI2), while the renal tubular cells generate PGE2. Investigations in vitro and in vivo have demonstrated that renin release is stimulated by prostaglandins and that the effect is due to a direct interaction with the JG-cells (Freeman et al. 1984). PGI2 acts to increase intracellular cAMP production through activation of IP receptors, while PGE2 produces different cellular responses depending on which receptor subtype (EP1 - EP4) that is expressed in the target cell. Jensen et al (1999) found that the EP4 receptor, which is coupled to cAMP production, was strongly expressed in glomeruli and in renin-secreting JG granular cells, and that the receptor expression was increased by salt deprivation. Consistent with this, PGE2-evoked cAMP production and renin secretion by JG cells from salt-deprived animals were significantly higher compared with cells obtained from salt-loaded animals. IP-receptor expression was not affected by salt intake.
COX-1 is expressed at high levels in collecting duct cells, interstitial cells, endothelial cells, and smooth muscle cells of pre- and postglomerular vessels in both humans, primates, rats and dogs (Khan et al. 1998, Komhoff et al. 1997), while COX-2 has been reported to be present in macula densa in several species (see above).
Lipoxygenase products (leukotriens) are also probably synthesized locally, and inhibitory effects on renin release of some of the metabolites, and a possible interplay with the cyclooxygenase products have been reported (Antonipillai 1990) but the possible relevance to the physiological control of secretion remains speculative.
The kallikrein-kinin system is active in the kidney. The end product, bradykinin, influences the vascular resistance, the blood pressure and kidney function in the opposite direction of that of the RAS. Biochemically, the two systems are closely linked since the same enzyme - ACE - is responsible for the generation of angII and the breakdown of bradykinin. Furthermore, kallikrein is capable of activating prorenin. Kallikrein has even been reported to stimulate renin release directly. The physiological interplay between these two systems is potentially important, but is still unresolved (Hackenthal et al. 1990).
The atrial natriuretic peptide (ANP) is a 28 amino acid peptide, which is produced by the heart and reaches the kidney by the blood stream. ANP activates membrane bound guanylyl cyclase after activation of membrane bound receptors and increases the production of cyclic GMP. Atrial natriuretic factor was reported to stimulate renin release from the isolated rat kidney (Hackenthal et al. 1985), have no effect on renin release from isolated rabbit afferent arterioles (Itoh et al. 1987) or from dispersed rat JG-cells and kidney slices (Takagi et al. 1988). Henrich et al. (1988) reported that ANP caused inhibition of renin secretion from rat cortical slices, and isolated juxtaglomerular cells. The ANP-treatment was associated with an increase in the generation of cGMP, but not cAMP. The apparent inconsistent findings may be resolved by the hypothesis that the stimulatory effect of cGMP is mediated through inhibition of PDE-3 and subsequent increase in the concentration of cAMP (Chiu and Reid, 1996, Kurtz et al. 1998), while the inhibition may be related to activation of cGKII (Wagner et al. 1998). Which physiological circumstances that mediate predominance of the stimulatory versus the inhibitory pathway are currently unknown.
CONCLUSIONS
Renal renin is synthesized, stored, and released from cells located in the afferent glomerular arteriole. The initial transcriptional product is preprorenin. Cleaving off the presegment leaves prorenin, which is glycosylated, and passes the Golgi apparatus. From here, it is either secreted immediately or stored in secretory granules where activation occurs by removal of the prosegment. The secretion of granules is regulated and takes place by exocytosis. Important intracellular signal molecules include calcium, which probably inhibits secretion, and cyclic AMP, which stimulates secretion. In addition, osmotic water movements also play a role.
The physiological regulation involves a complex interplay that involves the following mechanisms
1) a baroreceptor: lowered perfusion pressure to the kidney stimulates renin release,
2) a tubular signal: decrease in Cl- concentration at the macula densa stimulates renin release
3) the nerve supply of the kidney, which stimulates directly via ß-receptors on the JG-cells, and also indirectly via alpha-adrenergic influence on hemodynamic and tubular resorption, and
4) local and circulating hormones, particularly angiotensin II, nitric oxide and prostaglandins.
Acknowledgments. This review is based on a paper published in 1993 (Skøtt & Jensen 1993). Work from the author's laboratory was supported by the Danish Medical Research Council, the NOVO Foundation, and he Danish Heart Association.