PANEL DE DISCUSION |
ENDOTHELINS IN RENAL TRANSPLANTATION
Saraladevi Naicker
University of Durban
Durban. South Africa
email: naickers17@MED.UND.AC.ZA
Renal transplantation is the treatment of choice for patients with end stage renal disease. Success in transplantation is limited by the availability of donor organs and the occurrence of complications such as acute rejection , infections and chronic allograft nephropathy. Renal allograft rejection depends on the co-ordinated activation of alloreactive T cells and antigen-presenting cells (e.g. monocyte-macrophages, dendritic cells and B cells). This involves the activity of antibodies and inflammatory mediators, adhesion molecules, chemokines and cytokines, including components of the complement system, coagulation factors, leukotrienes and kinins. Cytokines also activate macrophages and other inflammatory cells.
Long term graft survival rate has not improved despite major improvements in one year graft survival. The half-life of cadaveric renal allografts remains at approximately 7 years (Paul and Benediktsson, 1993). The major reason for graft loss now is chronic rejection or chronic allograft nephropathy, which may be related to poor HLA matching, more frequent acute rejection episodes, cytomegalovirus infections, ischaemic and reperfusion injury to the graft, the initial amount of functioning renal mass and nephron number.
ENDOTHELINS
Yanagisawa et al. (1988) reported an endothelium-derived factor (a 21 amino acid peptide), which they named endothelin(ET), as the most potent vasoconstrictor described to date. Subsequent studies showed that ET is one of a family of 3 isopeptides. ET-1 is the major isopeptide produced by human endothelial cells and is present in the greatest concentration in blood. The concentrations of ET-1, though detectable in the human circulation, are very low (in the picomolar range), probably reflecting spill-over from local ET release. However, as ET-1 is released predominantly in an abluminal direction towards the underlying smooth muscle (Wagner et al., 1992), the tissue concentration is likely to be sufficiently high to activate local receptors.
Several agents, including insulin, thrombin, low density lipoprotein, angiotensin II, vasopressin and ET-1 itself (Emori et al., 1991; Boulanger et al., 1992; Emori et al., 1992; Kohno et al., 1992; Benatti et al., 1994) enhance ET-1 generation via activation of protein kinase C (PKC). Synthesis of ET-1 is inhibited by thrombin (Boulanger and L³scher, 1990), heparin (Yokokawa et al., 1993), atrial and brain natriuretic peptides (Kohno et al., 1991) and by the prostanoids prostaglandins E2 and prostacyclin (Prins et al., 1994). In the human genome, the ET-1 gene is found on chromosome 6 (Bloch et al., 1989; Hoehe et al., 1993), the ET-2 gene on chromosome 1 (Bloch et al., 1991) and the ET-3 gene on chromosome 20 (Bloch et al., 1989). ET gene expression has been demonstrated in the brain and spinal cord, lung, kidney, gut, eye, pituitary and amnion (reviewed by Simonson, 1993). The endothelium is the major site of ET gene expression. Glomerular immunoreactive ET-1 is localised primarily in the endothelium and to a lesser extent, in the mesangium. The proximal location of cells in the cortex and medulla expressing ET binding sites and preproET mRNA transcripts suggests that ET acts as an autocrine or paracrine peptide (Simonson, 1993).
ENDOTHELIN RECEPTORS
The ETA type receptor is characterised by its very high (subnanomolar) affinity for ET-1 and ET-2 and its 70-100 fold lower affinity for ET-3, while the ETB receptor has high and equal affinity for all 3 isopeptides. The cDNAs encoding the human ETA and ETB receptors predict 427 and 442 aminoacids respectively.
The ETA and ETB receptor genes, located on chromosomes 4 (Hosada et al., 1992) and 13 (Arai et al., 1993) respectively, have similar structural organisation. Exogenous factors can act through these regions to increase receptor transcription, namely upregulation of ET receptor mRNA by insulin (Frank et al., 1993), or ETB receptor mRNA by angiotensin II (Kanno et al., 1993). In vascular tissue, ETA receptor mRNA is expressed predominantly in smooth muscle, while ETB receptor mRNA is most abundant in endothelial cells, suggesting that constriction of vascular smooth muscle is mediated predominantly by ETA receptors, and that constriction is modified by release of relaxing factors from the endothelium via stimulation of ETB receptors (reviewed by Gray and Webb, 1996).
ENDOTHELIN AND RENAL PHYSIOLOGY
Endothelins affect three major aspects of renal physiology: vascular and mesangial tone, cell proliferation and matrix formation and sodium and water excretion.
Renal vascular tone is regulated by (-and (-adrenergic sympathetic output, and by the levels of circulating vasoactive compounds such as angiotensin II, prostaglandins, arginine vasopressin, atrial natriuretic peptide and kinins. Renal vasomotor tone is exquisitely sensitive to ET (Simonson, 1993). ET-1 may induce renal vasoconstriction via platelet-activating factor and increase in cytosolic free calcium (Simonson and Dunn, 1992). On a molar basis, ET-1 is 30 times more potent than angiotensin II and 50 times more potent than noradrenaline (Cairns et al., 1989).Secretion of ET by endothelial cells would counteract the vasodilatory signals released by endothelial cells, such as endothelin derived relaxing factor/nitric oxide and prostacyclin.
EFFECT ON THE MESANGIUM
Mesangial cells synthesize and assemble the mesangial matrix. Endothelins have both contractile and pro-mitogenic actions on mesangial cells, which might contribute to the glomerular response to injury. ET-1 is a potent growth factor for mesangial cells and stimulates quiescent mesangial cells to enter G1 and proliferate. Proinflammatory agents such as IL-1 and TGF( stimulate ET secretion in endothelial and mesangial cells, supporting this hypothesis.
SODIUM HANDLING
ET may regulate extracellular fluid volume and mean arterial pressure. ET appears to have diverse effects on renal sodium handling and the precise role of ET in renal sodium excretion is unclear. Systemic infusions of ET-1 decrease sodium excretion in some studies (Goetz et al., 1988; Hirata et al., 1989; Miller et al., 1989) in but others, (King et al., 1989; Garcia et al., 1990) ET-1 was modestly natriuretic despite a decline in renal blood flow (RBF). ET reduces GFR and would therefore be expected to reduce the filtered load of sodium, resulting in decreased sodium excretion.
ENDOTHELIN AND WATER REABSORPTION
Renal water reabsorption is regulated indirectly by plasma osmolality, which evokes changes in pituitary arginine vasopressin release. ET increases urine flow rate despite a decrease in RBF and GFR (Goetz et al., 1988; Badr et al., 1989), suggesting that ET may regulate water reabsorption. ET secretion, stimulated by water deprivation, could reduce water excretion directly by inhibiting baroreceptor reflexes or reducing GFR.
ENDOTHELIN AND RENAL TRANSPLANTATION
Plasma ET levels were reported to be elevated in predialysis patients with chronic renal failure and in patients on regular haemodialysis; plasma ET levels were normal in stable renal transplant patients treated with cyclosporine A (Stockenhuber et al., 1992). Chronic renal failure and hypertension
Alterations in ET-1 in the renal vasculature and renal tubules have differing effects on blood pressure. In the vasculature, increases in ET-1 predominantly cause vasoconstriction with a hypertensive effect. Increased ET-1 in the nephron probably enhances sodium and water excretion, favouring hypotension (reviewed by Markewitz and Kohan, 1995; Schiffrin, 1995).
Plasma ET-1 levels are frequently elevated in chronic renal failure or dialysis patients (Shichiri et al., 1990;Warrens et al., 1990; Stockenhuber et al., 1992). Further fractionation by gel permeation chromatography revealed borderline increased plasma ET-1 with marked elevations of big ET-1 and other large forms of ET, with an increase of about 500% above control (Saito et al., 1991). Plasma ET-1 levels may correlate with the degree of hypertension in chronic renal failure patients, though this has not been uniformly noted. The kidney is a major site of ET-1 metabolism (Abassi et al., 1992); the plasma ET-1 half-life is increased in bilaterally nephrectomised rats (Kohno et al, 1989). Dialysis may elevate ET-1 levels. The type of dialysis membrane may influence plasma ET-1 levels (Niwa et al., 1993; Ross et al., 1993); plasma ET-1 levels are reduced when high flux membranes are used. Thus patients with chronic renal failure or end stage renal disease have borderline elevated plasma ET-1 levels due partly to reduced ET-1 clearance while intrarenal generation of ET-1 may be increased.
Cyclosporine nephrotoxicity
Endothelin-1 may be involved in the pathogenesis of many toxic nephropathies, including that due to cyclosporine A (CyA). An association between increased plasma ET levels and CyA was first suggested in a renal transplant patient receiving high doses of CyA (Fogo et al., 1990). Endothelin-1 mediates acute CyA-induced renal vasoconstriction. Cyclosporine and related immunosuppressants (eg FK506) directly stimulate ET-1 release from mesangial/or endothelial cells (Langman et al., 1994; Goodall et al., 1995; Kohno et al., 1995). Cyclosporine A increases renal ET-1 mRNA expression (Iwasaki et al., 1994). Anti-ET antibodies and ET receptor antagonists ameliorate acute CyA-induced renal vasoconstriction (Kon and Awezu, 1992; Lanese and Conger, 1993; Conger et al., 1994; Brooks and Contino, 1995; Kon et al., 1995).
Salt-depleted rats given CyA daily for 5 weeks developed a reduced GFR and tubulointerstitial fibrosis. The decrease in GFR was ameliorated by concurrent administration of an ETA/ETB receptor antagonist, while fibrotic changes were unaltered (Kon et al., 1995).
Acute rejection
Plasma ET levels were normal in stable renal transplant patients treated with CyA (Stockenhuber et al., 1992). After administration of CyA, plasma ET levels increase transiently in transplant patients because ET is rapidly metabolised and cleared from the systemic circulation (Grieff et al., 1993).
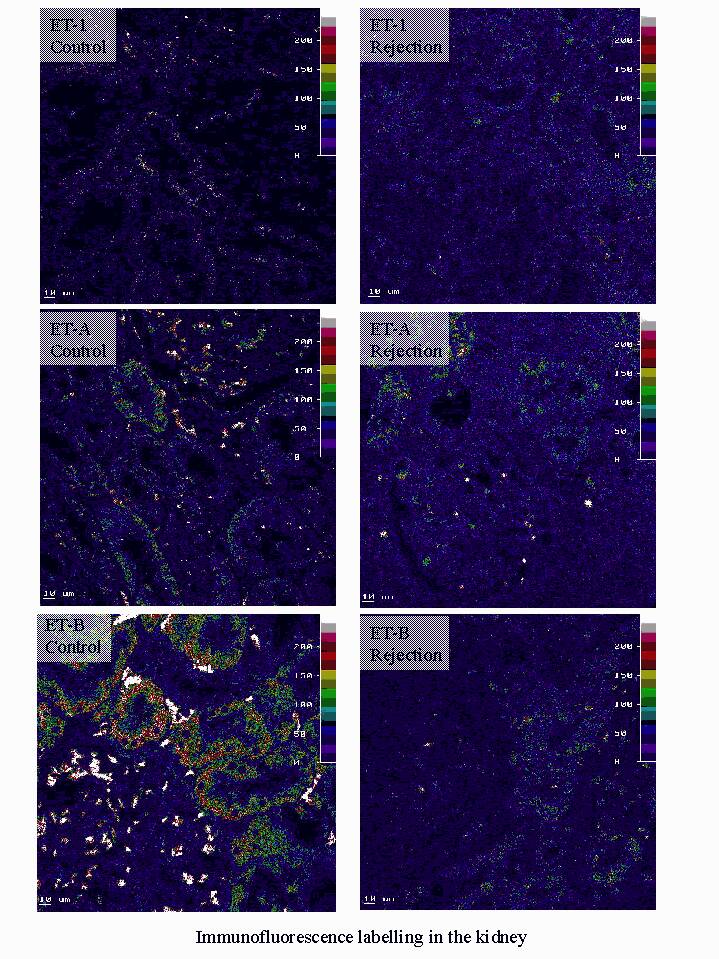
Plasma ET-1 showed a small but significant increase in acute rejection compared to normal controls; while ETA receptor immunoreactivity was similar to that of control kidney, ETB receptor immunolabelling was increased in glomeruli and decreased in distal tubules in kidney biopsies with acute rejection (Naicker et al., 1999).
The inflammatory process of acute cellular rejection is predominantly confined to the interstitium and marked intragraft upregulation of ET-1 may occur without significant changes in ET-1 plasma concentrations (Watschinger et al., 1995), unless there is endothelial damage as occurs with vascular rejection ( Watschinger et al., 1994).
Activated mononuclear cells that infiltrate the allograft secrete a variety of cytokines (Blancho et al., 1993; Halloran et al., 1993; Hancock 1984), that have been shown to influence the production of ET-1 by other cells in vitro. Tumour necrosis factor ( increases ET-1 mRNA and ET-1 release in capillary endothelial cells, epithelial cells and rat mesangial cells (Kohan 1991); IL-1( induces ET-1 release from renal epithelial cells (Ohta et al., 1990). Platelet activation and intravascular coagulation (attributable to endothelial injury) appear to trigger the release of TGF(, thromboxane A2, PDGF and thrombin from platelets accumulating in the graft. This results in ET-1 secretion and upregulation of ET-1 gene expression in endothelial, vascular smooth muscle and renal mesangial and epithelial cells (Kurihara et al., 1989; Watschinger and Sayegh, 1996).
Chronic rejection/chronic allograft nephropathy
Renal allografts with chronic rejection and transplant-associated arteriosclerosis were reported to express 6-fold more ET-1 in the neointima of the vasculature, when compared to allografts with acute rejection or normal control kidneys (Simonson et al.,1998). A strong correlation was noted between vascular ET-1 expression and hypertension in patients with chronic rejection.
Periglomerular and perivascular macrophages secrete cytokines that are profibrogenic, including platelet-derived growth factor (PDGF) and transforming growth factor-( (TGF(). This is associated with upregulation of the ET-1 gene expression in endothelial, vascular smooth muscle and renal mesangial and epithelial cells and results in ET-1 secretion (Kurihara et al., 1989; Watschinger and Sayegh,1996).
Endothelin-1 may also contribute to excessive accumulation of extracellular matrix components and fibrosis by increasing renal cell fibronectin and collagen production, tissue inhibitor of metalloprotease levels and the release of cytokines that stimulate matrix accumulation (Ong et al., 1994; Ruiz-Ortega et al., 1994). ET-1 antagonism decreases matrix accumulation in experimental models of glomerulonephritis (Fukuda et al., 1995). Chronic treatment with an ET receptor antagonist attenuates increases in glomerular mRNA levels of collagen, laminin, tumor necrosis factor , TGF-(, PDGF and basic fibroblast growth factor in diabetic rats (Nakamura et al., 1995). Once substantial renal scarring occurs, there is an inevitable progression to end stage kidney disease, a process involving gradual glomerular sclerosis and interstitial fibrosis. Endothelin receptor blockade reduced proteinuria and glomerulosclerosis and protected against hypertension and elevations in serum creatinine in the 5/6 nephrectomy rat model (Benigni et al., 1993 and 1996).The gradual functional deterioration caused by the development of glomerulosclerosis and arterial obliteration may also cause systemic hypertension which causes the remaining functional glomeruli to hyperfilter before eventually fibrosing, thus resulting in progressive renal damage (Neuringer and Brenner, 1992).
REFERENCES
{kind=link}