
|
Paneles de Discussión
Paneais de Discussio Comunicaciones libres
Comunicaçoes livres |
New insights into the natural history of IgA nephropathy in adults.
Colin C. Geddes FRCP. (Gla)
Introduction IgA nephropathy (IgAN) was first described in 1969 [1] and is now recognised as the commonest primary glomerulopathy. The long-term outcome is highly variable [2]. Knowledge about the natural history of IgAN provides prognostic information, informs decisions about the risks and benefits of therapeutic options, can refine the design of clinical trials and enhances our understanding of the pathophysiology of progressive renal disease. This review will consider questions patients might ask and highlight recently published studies that have added to our knowledge about the natural history of IgAN. Clinical course of IgAN Reports from post mortem examinations and patients with recurrent IgAN in a renal transplant suggest that the hallmark of IgAN, mesangial deposition of IgA immune complexes, may be associated with a period of no clinical manifestation. At least a proportion of patients will subsequently develop microscopic haematuria which may be accompanied by proteinuria and hypertension. It is not known if, given enough time, all patients with mesangial IgA deposition would develop haematuria and proteinuria. Of the patients who develop proteinuria and hypertension a proportion will develop impaired renal function which is usually progressive. The time from first clinical manifestation to development of impaired renal function is variable and the rate of progression of renal failure is also variable. It is not known why or when the process that results in mesangial IgA deposition also results in haematuria and proteinuria. The pathological process that leads to progressive renal failure shares many features with other glomerular diseases i.e. persistent proteinuria and glomerular hypertension leading to progressive tubular atrophy, interstitial fibrosis and glomerulosclerosis. Thus the natural history for the majority of patients with IgAN can be represented in the figure below 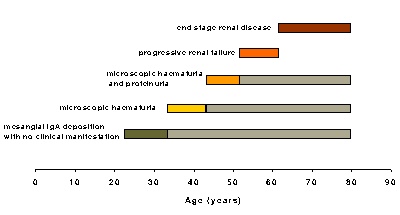
Other clinical patterns of IgAN The classical description of episodes of transient painless macroscopic haematuria occurring at the same time as upper respiratory tract infections in young men happens in only a minority of patients and usually has a benign prognosis (see below). More rarely still patients can experience acute renal failure associated with a crescentic form of IgAN, acute renal failure during periods of macroscopic haematuria (related to acute tubular necrosis caused by haemoglobin tubular toxicity) or nephrotic syndrome. Diagnosis of IgAN When considering the natural history of IgAN it is important to appreciate that the diagnosis of IgAN can only be made by histological examination of renal cortex that includes glomeruli. Whether or not a patient with IgAN has a renal biopsy to make the diagnosis is clearly dependent on several factors: 1) the patient may have no contact with a doctor until macroscopic haematuria develops or a urine dipstick test is performed (e.g. at an employment medical screening programme); 2) the identification of microscopic haematuria and proteinuria, (or even renal impairment) by a doctor may not necessarily result in referral to a nephrologist; 3) if the patient does attend a nephrologist a renal biopsy may not be considered justified; 4) even if a renal biopsy is performed there may be no glomeruli in the piece of tissue prepared for immunofluorescence. Incidence of IgAN Published reports of the natural history of biopsy proven IgAN therefore contain only an unquantifiable proportion of the patients in the general population who have IgAN and cannot be used to determine the incidence of IgAN. In a mass screening programme of 50,501 Japanese working men 772 were found to have urine dipstick abnormalities and the incidence of IgA nephropathy was 143 per million per year [3]. This is likely to be an underestimate, however, as not all patients with urine dipstick abnormalities had a renal biopsy. The incidence of end-stage renal disease (ESRD) due to IgA nephropathy in USA between 1996 and 2000 was 2.54 per million population (including children) [4]. This figure is also likely to be an underestimate because of patients with IgAN who do not get a histological diagnosis and are labelled as "cause unknown". Most studies report more males than females with IgAN. It is not clear if this is because there is a true gender difference in incidence or if females are less likely to be referred for investigation. Microscopic haematuria may be more likely to be attributed to other causes such as urine tract infection or menstruation in females. IgAN appears to be more common in subjects of Asian and Caucasian descent than black descent. What does the patient want to know? From the outline of the natural history described a patient might have several questions:
I will discuss each of these questions in turn and demonstrate how the limitations of the published data restrict the precision of the answers. Why did I get IgAN? A detailed discussion of the aetiology of IgA is beyond the scope of this review. Suffice to say that the observation that conditions such as Henoch Schonlein purpura, gluten enteropathy, hepatic cirrhosis, HIV infection and familial mutations of a locus on chromosome 6 (6q22-23) can be associated with mesangial deposition of IgA implies that the deposition of IgAN may be a common feature of several different pathological processes. How long have I had IgAN? This question is usually impossible to answer since, as shown in figure 1, the period between the first deposition of IgAN and the clinical manifestations is impossible to define. In the absence of regular screening, the time to the identification of the first clinical manifestation is dependent on chance testing of the urine or blood. Will I always have IgAN? Most patients with microscopic haematuria and asymptomatic proteinuria will continue to have abnormal urinalysis during long-term follow up. Alamartine reported repeat renal biopsies performed at five years in 73 patients with persistent proteinuria but a normal or near-normal initial plasma creatinine concentration and found that histological improvement occurred in only 4% [5]. In patients with IgAN who subsequently receive a renal transplant approximately half will experience recurrent mesangial IgA deposition in the transplant indicating on-going disease activity, usually many years after onset. Will I need dialysis? This is probably the most important question for the future health of the patient. Being able to identify who will need dialysis is also important as these are the patients who will gain most from therapeutic intervention.
To answer the question the published literature must be applied in the context of the patient’s clinical features. D’Amico found that 10 year actuarial renal survival varied from 57%-94% in 18 studies from around the world [2]. In 30 studies increased serum creatinine at presentation and increasing proteinuria at presentation were strong predictors of future need for dialysis, increasing arterial blood pressure was an intermediate predictor while increasing age, male gender and absence of a history of macroscopic haematuria were weak predictors. Rauta et al recently showed that in patients with normal renal function at presentation, the number of urinary erythrocytes were also predictive of outcome [7]. Studies with low renal survival tended to include patients with poorer renal function and higher proteinuria at presentation and so it is not clear if geographic variability is entirely explained by varying referral and biopsy practices. We recently published the long term natural history of the largest cohort of patients with IgAN reported to date drawn from four centres in three continents (Glasgow, UK; Helsinki, Finland; Sydney, Australia; Toronto, Canada) [8]. Referral and biopsy practice varied between centres with patients with isolated microscopic haematuria being more likely to be referred to a nephrologist and have a renal biopsy in Helsinki and Sydney than Glasgow and Toronto. 10 year renal survival of the 711 patients ranged from 93.3% in Helsinki to 61.4% in Toronto. Studies using renal survival as an end-point are limited by the fact that a patient who has a rise in serum creatinine during follow up from 100μmol/L to 500μmol/L is counted as the same as a patient with stable renal function. To overcome this both renal survival and slope of the creatinine clearance were analysed and we found that poor renal function at presentation and increased proteinuria at presentation were adverse risk factors for both end-points. When multivariate analysis was applied the centre effect was greatly diminished suggesting that most of the variation in outcome was due to the variation in referral and biopsy practices. Furthermore there was no gender effect and advancing age was associated with slower deterioration in renal function. Many studies have shown that glomerulosclerosis and tubulo-interstitial scarring also predict poor outcome but it is not clear if these add to the predictive power of the clinical data [2; 9]. In recent years there has been a proliferation of reports of various gene polymorphisms being predictive of disease severity. However when studies of gene polymorphisms have been repeated in other centres the results have usually been non-confirmatory. A recent report of increased body weight being an independent risk factor for progression also requires confirmation [10] Few studies have attempted to predict individual risk of renal failure in patients with IgAN. It seems unlikely that models based on a snapshot of a few clinical features from the time of diagnosis in a complex chronic disease will be able to predict outcome with accuracy. Bartosik et al have recently shown that the accuracy of a predictive model improves when blood pressure and proteinuria during the early years of follow up are included [9]. We advise patients that approximately 30% of patients identified in our practice will progress to ESRD in the 10 years after diagnosis. If the patient has <0.5g urine protein excretion per day and serum creatinine is <110μmol/L then they are told their risk is lower than average and if urine protein excretion is >2g/day and serum creatinine >150μmol/L then they are told their risk is higher than average. We emphasize that these predictions are imprecise, cannot predict what will happen after 10 years and do not take in to account the likely benefit of recent advances in treatment including lowered blood pressure targets and widespread use of inhibitors of the renin-angiotensin system. Will I need follow-up by a nephrologist? Recently published data from Hong Kong from 72 consecutive patients with histologically confirmed IgA nephropathy who presented with haematuria and minimal proteinuria (0.4 g/day or less), were normotensive and had normal renal function at presentation show that during a median of 7 years follow up 24 (33%) patients developed proteinuria of 1 g per day or more, and 5 (7%) developed impaired renal function [11]. These data suggest that even patients with isolated microscopic haematuria should have regular measures of proteinuria, blood pressure and serum creatinine. Whether follow up should be done by a nephrologist will depend on local resources. Conclusion Many questions about the natural history of IgAN remain unanswered. It seems sensible to focus research efforts on why patients with microscopic haematuria and proteinuria progress to impaired renal function and what effect modern treatment has on the clinical course of IgAN. References 1. Berger J: IgA glomerular deposits in renal disease. Transplantation Proceedings 1969; 1:939-944 2. D’Amico G. Natural history of idiopathic IgA nephropathy: role of clinical and histological prognostic factors. Am J Kidney Dis. 2000; 36:227-37. 3. Yamagata K, Takahashi H, Tomida C, Yamagata Y, Koyama A. Prognosis of asymptomatic hematuria and/or proteinuria in men. High prevalence of IgA nephropathy among proteinuric patients found in mass screening. Nephron. 2002; 91:34-42. 4. U.S. Renal Data System, USRDS 2002 Annual Data Report: Atlas of End-Stage Renal Disease in the United States, National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases, Bethesda, MD, 2002.* 5. Alamartine E; Sabatier JC; Berthoux FC. Comparison of pathological lesions on repeated renal biopsies in 73 patients with primary IgA glomerulonephritis: value of quantitative scoring and approach to final prognosis. Clin Nephrol 1990; 34:45-51. 6. Odum J; Peh CA; Clarkson AR; Bannister KM; Seymour AE; Gillis D; Thomas AC; Mathew TH; Woodroffe AJ. Recurrent mesangial IgA nephritis following renal transplantation. Nephrol Dial Transplant 1994; 9:309-12. 7. Rauta V, Finne P, Fagerudd J, Rosenlof K, Tornroth T, Gronhagen-Riska C. Factors associated with progression of IgA nephropathy are related to renal function--a model for estimating risk of progression in mild disease. Clin Nephrol. 2002; 58:85-94. 8. Geddes CC, Rauta V, Gronhagen-Riska C, Bartosik LP, Jardine AG, Ibels LS, Pei Y, Cattran DC. A tricontinental view of IgA nephropathy. Nephrol Dial Transplant. 2003; 18:1541-8. 9. Bartosik LP, Lajoie G, Sugar L, Cattran DC. Predicting progression in IgA nephropathy. Am J Kidney Dis. 2001; 38:728-35. 10. Bonnet F, Deprele C, Sassolas A, Moulin P, Alamartine E, Berthezene F, Berthoux F.Excessive body weight as a new independent risk factor for clinical and pathological progression in primary IgA nephritis. Am J Kidney Dis. 2001; 37:720-7. 11. Szeto CC, Lai FM, To KF, Wong TY, Chow KM, Choi PC, Lui SF, Li PK. The natural history of immunoglobulin a nephropathy among patients with hematuria and minimal proteinuria. Am J Med. 2001; 15;110:434-7. * The data reported here have been supplied by the United States Renal Data System (USRDS). The interpretation and reporting of these data are the responsibility of the author(s) and in no way should be seen as an official policy or interpretation of the U.S. government |