
|
Paneles de Discussión
Paneais de Discussio Comunicaciones libres
Comunicaçoes livres |
Cytokines in the pathogenesis and treatment of experimental crescentic glomerulonephritis Frederick W. K. Tam MB BChir PhD MRCP
Renal Section, Division of Medicine, Faculty of Medicine, Imperial College London, Charing Cross and Hammersmith Hospitals, London f.tam@imperial.ac.uk Abstract Crescentic glomerulonephritis is a very aggressive form of glomerulonephritis. Cytokines are important in cell mediated immunity, in migration of monocytes/macrophages and T cells, and in inflammation. The role of cytokines in these processes has been demonstrated in rodent models of crescentic glomerulonephritis. The development of cell mediated immunity can be inhibited by suppressing Th1 responses with Th2 cytokines, such as interleukin-4 (IL-4). The severity of crescentic glomerulonephritis has been reduced successfully by blockade of cytokines using neutralising antibodies, recombinant a cytokine receptor antagonist and soluble cytokine receptors. Alternatively, synthesis of cytokines can be suppressed with lower molecular weight inhibitors (e.g. type IV phosphodiesterase inhibitor). Macrophage mediated inflammation can be suppressed by anti-inflammatory cytokines (e.g. IL-4, IL-11). Cytokines are realistic therapeutic targets in crescentic glomerulonephritis. There are ongoing clinical trials with anti-TNF- INTRODUCTION Crescentic glomerulonephritis, the most severe form of glomerulonephritis, may progress to renal failure rapidly. Current therapy consists of the administration of non-specific immunosuppressive drugs, which are associated with significant side-effects. Understanding of the role of cytokines has provided more specific targets for therapeutic intervention. Cytokines are involved in both cell mediated immunity and inflammation during pathogenesis of crescentic glomerulonephritis. CELL MEDIATED IMMUNITY Monocytes/macrophages and T cells are important in the pathogenesis of crescentic glomerulonephritis1. Experimental models are particularly useful in identifying the cytokines involved in the cell mediated immunity in the pathogenesis of crescentic glomerulonephritis. Using a knockout approach with a murine model of crescentic glomerulonephritis, Tipping and colleagues have demonstrated that interferon- The role of IL-18 is more complicated. Although IL-18 mRNA was detected from the glomeruli of WKY rats with nephrotoxic nephritis, expression of IL-18 was not detected. This may be due to the existence of a specific translational inhibitor4. Although direct involvement of IL-18 has not been reported, injection of recombinant IL-18 exacerbates crescentic glomerulonephritis in both wild type C57BL/6 mice and IL-12 knockout mice 4 and nephrotoxic nephritis in WKY rats5. The relative contribution of bone marrow derived cells and intrinsic renal cells in cytokines dependent cell mediated immunity was studied by bone marrow reconstitution of C57BL/6 mice (see Table 1). Table 1: the relative contribution of bone marrow (BM) derived cells and intrinsic renal cells in the pathogenesis of experimental crescentic glomerulonephritis.
Although these results seem to be very predictable from the current understanding of cytokines in development of T help (Th) subsets, there are several interesting conclusions from these studies. First, cell mediated immunity is critical in crescent formation although these models were induced by antibody against native or planted antigens of glomerular basement membrane. Second, deficiency of one cytokine (e.g. IL-12) may be overcome by systemic administration of another cytokine (e.g. IL-18). Third, although multiple cytokines are involved in pathogenesis of crescentic glomerulonephritis, knockout of a single cytokine is sufficient enough to reduce of crescent formation significantly. ROLE OF CHEMOKINES (Figure 1) 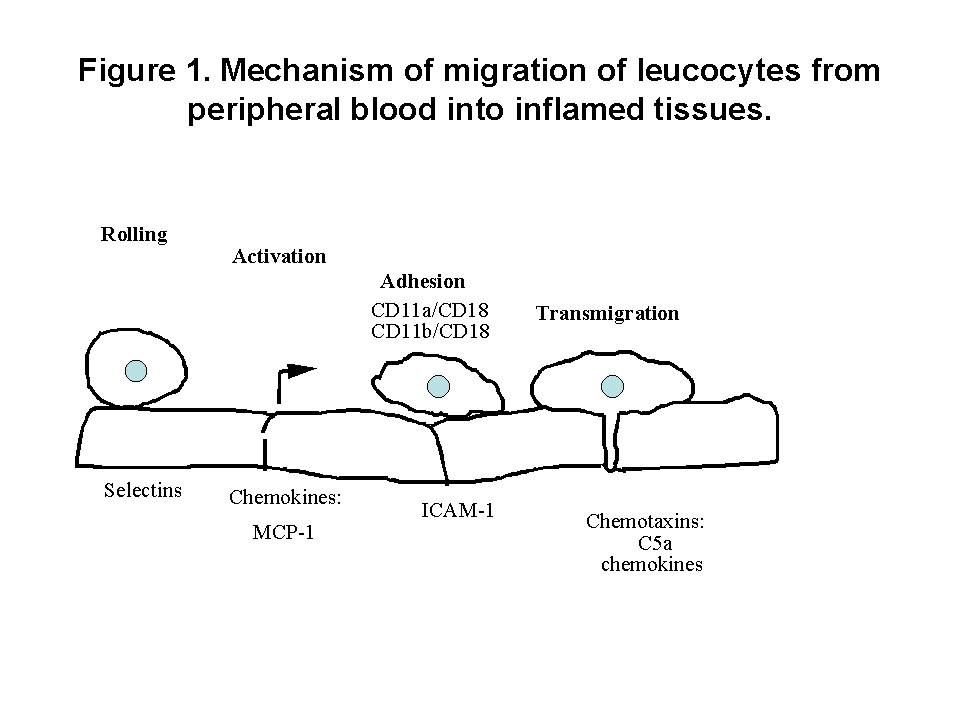 Recruitment of monocytes/macrophages and T cells into glomeruli is a critical event in pathogenesis of crescentic glomerulonephritis1. Expression of MCP-1 correlated with monocyte/macrophage influx into the glomeruli during the early stage of accelerated nephrotoxic nephritis in SD rats (Figure 2)7. Administration of antibodies to MCP-1 reduced crescent formation8. Increased expression of other chemokines has also been detected in experimental crescentic glomerulonephritis:
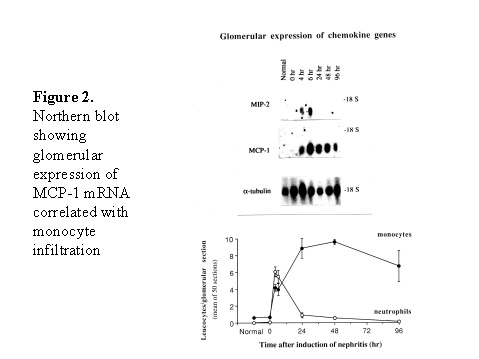 These chemokines are known be have roles in recruitment of macrophage and T cells. Administration of antibody to fractalkine receptor reduced macrophage infiltration and crescent formation9. However, chemokines may have complicated roles in vivo. Blockade of RANTES reduced cell infiltration but not the number of crescent formed10. A more recent study showed that RANTES antagonist aggravated experimental glomerulonephritis despite reduction of leucocyte infiltration11. Definite studies of other chemokines have not been reported yet. INFLAMMATION IL-1 and TNF- Interleukin-1 and TNF- Both IL-1 and TNF- As these cytokines have such powerful roles in glomerular inflammation, tight regulation is essential. This is best understood in IL-1 (Figure 3). The effect of IL-1 is partly regulated by the existence of a natural competitive receptor antagonist (IL-1RA). IL-1 is regulated further by the existence of two types of cell surface receptors. Type I IL-1 receptor (80kD) is the signalling receptor with long cytoplasmic tail. Type 2 IL-1 receptor (68kD), also known as IL-1 decoy receptor, only has a short cytoplasmic tail and it is unable to transduce signal. The extracellular portion of IL-1 type 2 receptor can also be shedded from cell surfaces. These soluble IL-1 receptors act as inhibitors to IL-1 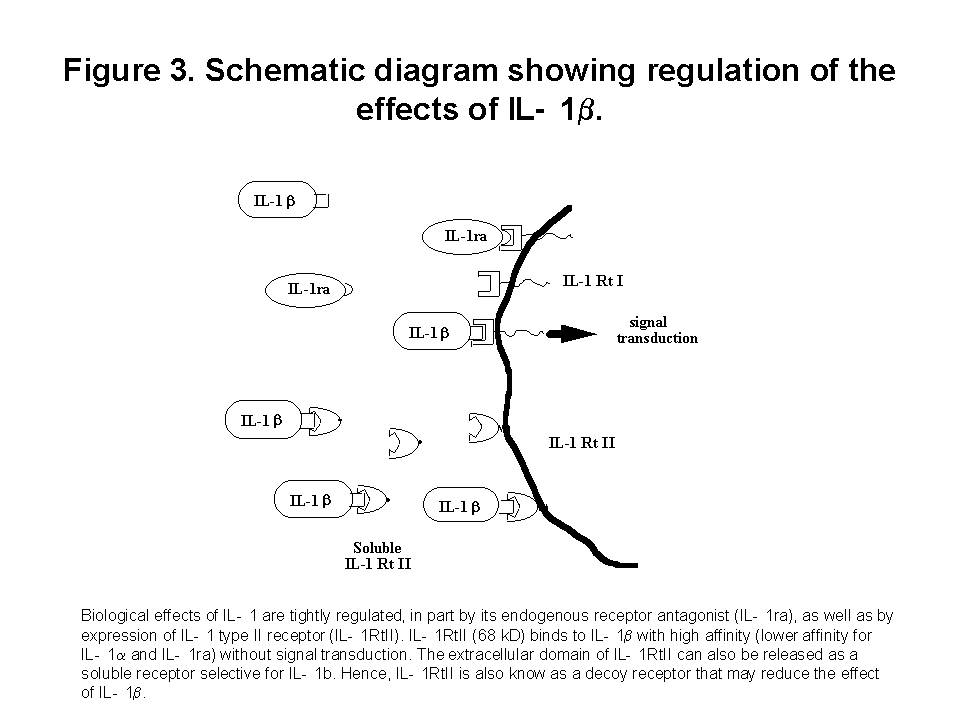 Co-ordinated glomerular expression of IL-RA and IL-1 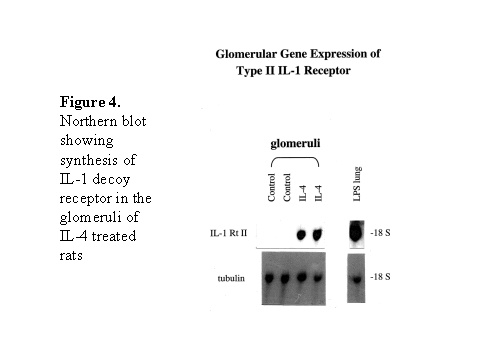 Severity of crescentic glomerulonephritis was reduced when pharmacological doses of recombinant IL-1RA was given14. Soluble IL-1 receptor has only been used in short term studies. Expression of IL-1 decoy receptor can be upregulated by IL-4. Systemic administration of IL-4 has been shown to upregulate glomerular expression of IL-1 decoy receptor in association with inactivation of macrophage and reduction of glomerular injury (Figure 5)15. 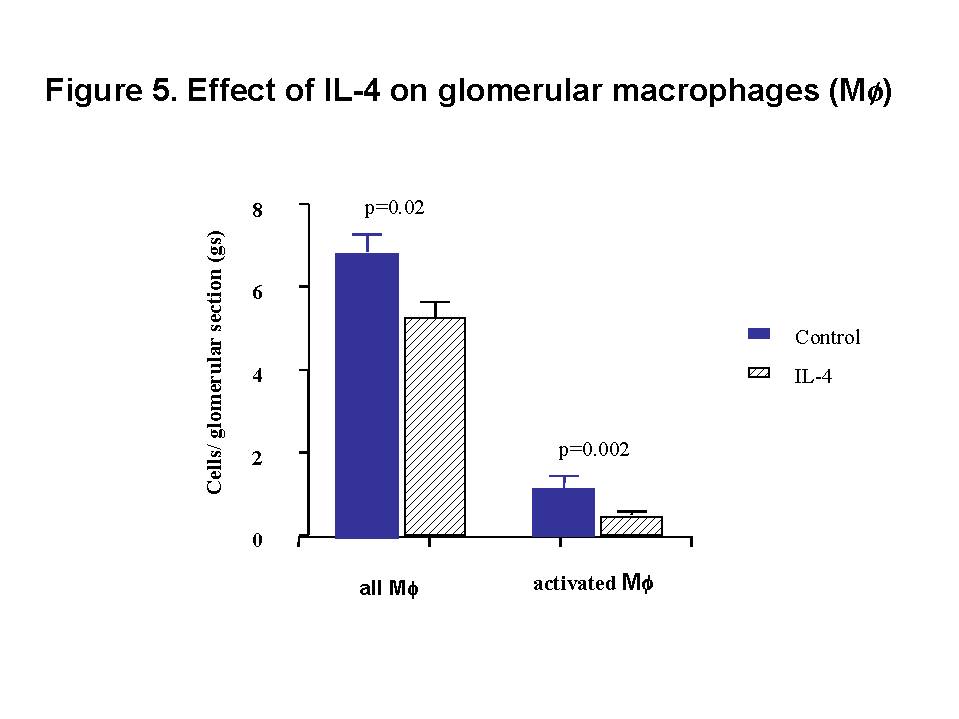 Administration of monoclonal antibody or soluble receptor to TNF- 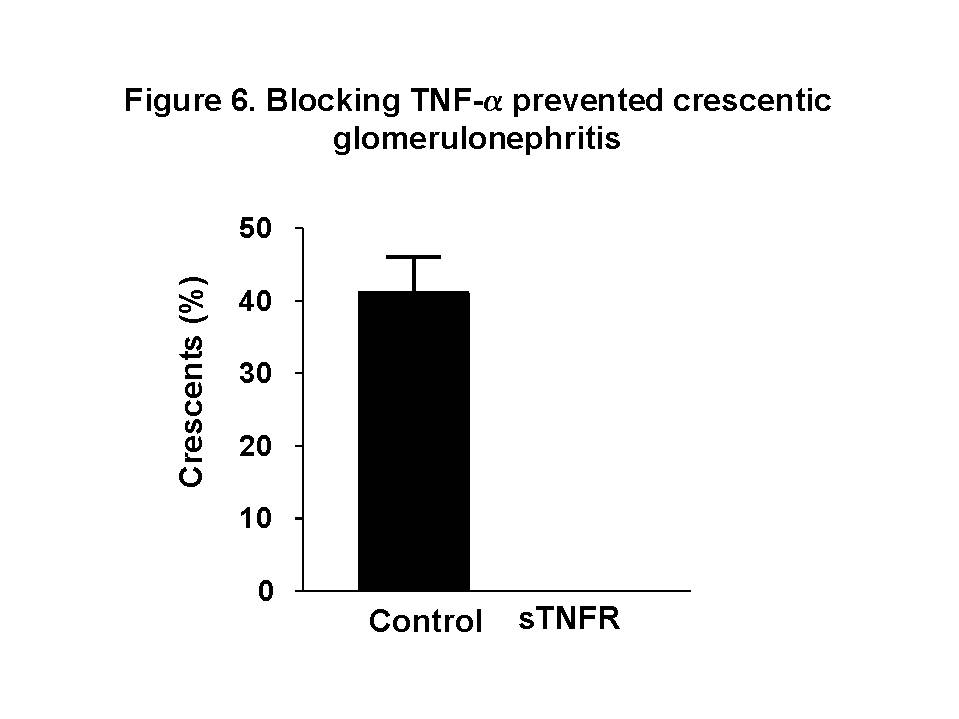 Synthesis of TNF- 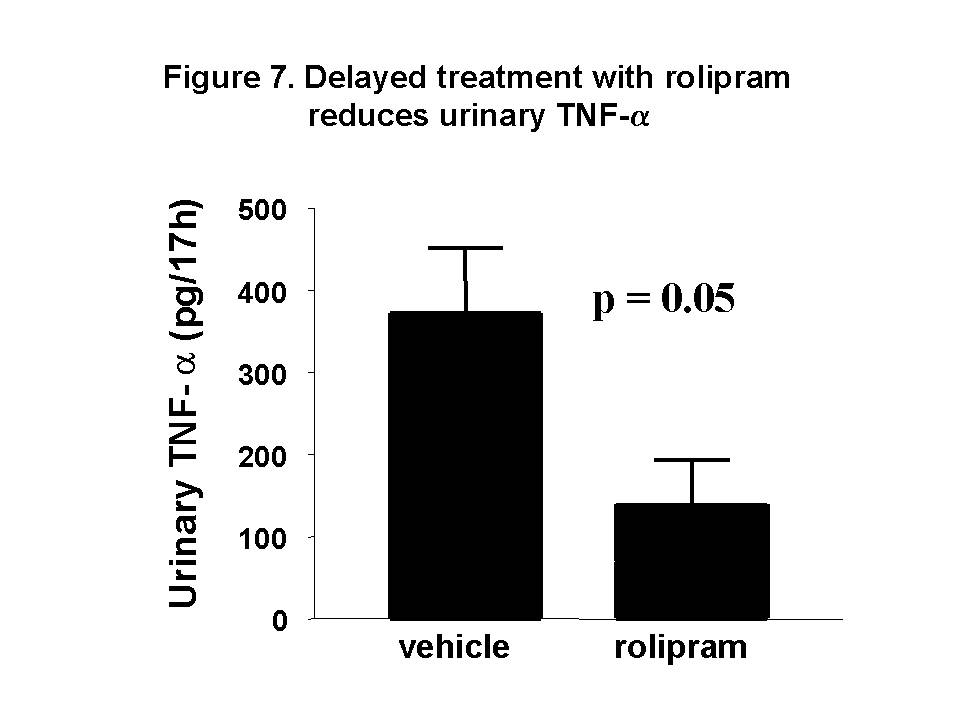 Systemic injection of rolipram, a type IV phosphodiesterase inhibitor, reduced crescent formation even when it was given after onset of nephritis. Initiation of treatment after onset of disease also reduced the severity of crescent formation (Figure 8)18. In accelerated nephrotoxic nephritis in C57BL/6 mice, glomerular macrophage and T cell numbers and also crescent formation were reduced significantly in TNF- 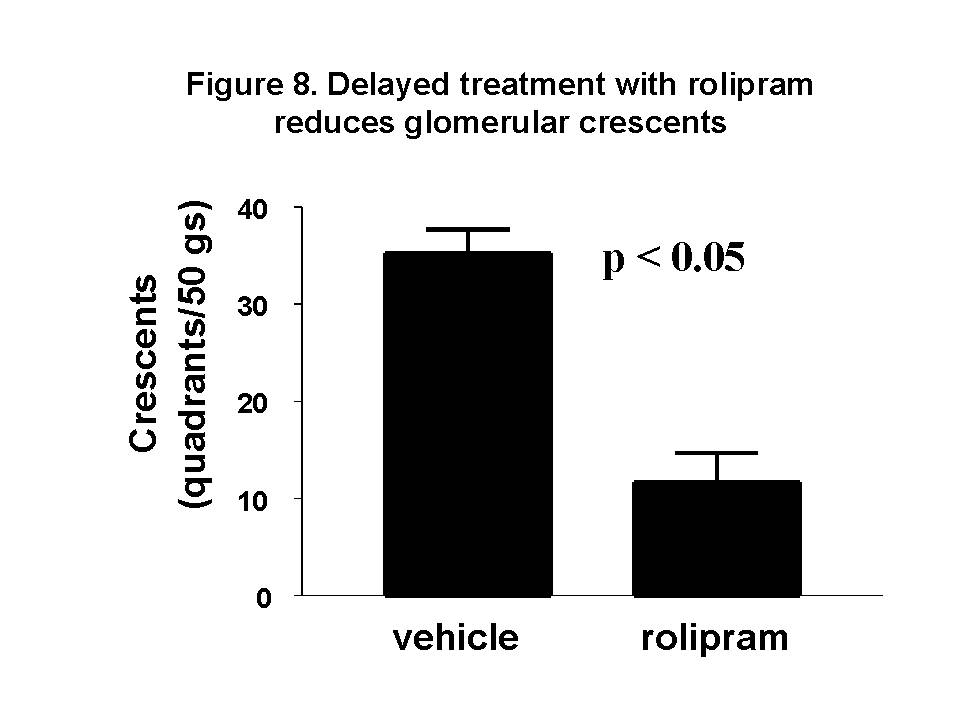 Interferon (IFN)- Macrophage activation is important in pathogenesis of crescentic glomerulonephritis. IFN- ANTI-INFLAMMATORY CYTOKINES There is a large family of cytokines that have anti-inflammatory effects. Some of them may also have a direct effect on T cell responses. It is important to understand the design of individual studies to distinguish whether it is inflammation, T cells responses or both being affected by the intervention. Interleukin-4 IL-4 is one of the most potent anti-inflammatory cytokines tested. As mentioned in the earlier section, systemic injection of IL-4 was shown to upregulate glomerular expression of IL-1 decoy receptor (Figure 4)15. IL-4 was still effective in reducing crescent formation when it was given after induction of nephritis19. A short treatment with IL-4 at an early stage of the disease resulted in a major reduction of injury even at day 28 (Figure 9). One of the important effects of IL-4 was inactivation of macrophages. The reduction in glomerulonephritis was probably due to reduction in total number of macrophages and the state of activation of macrophages. 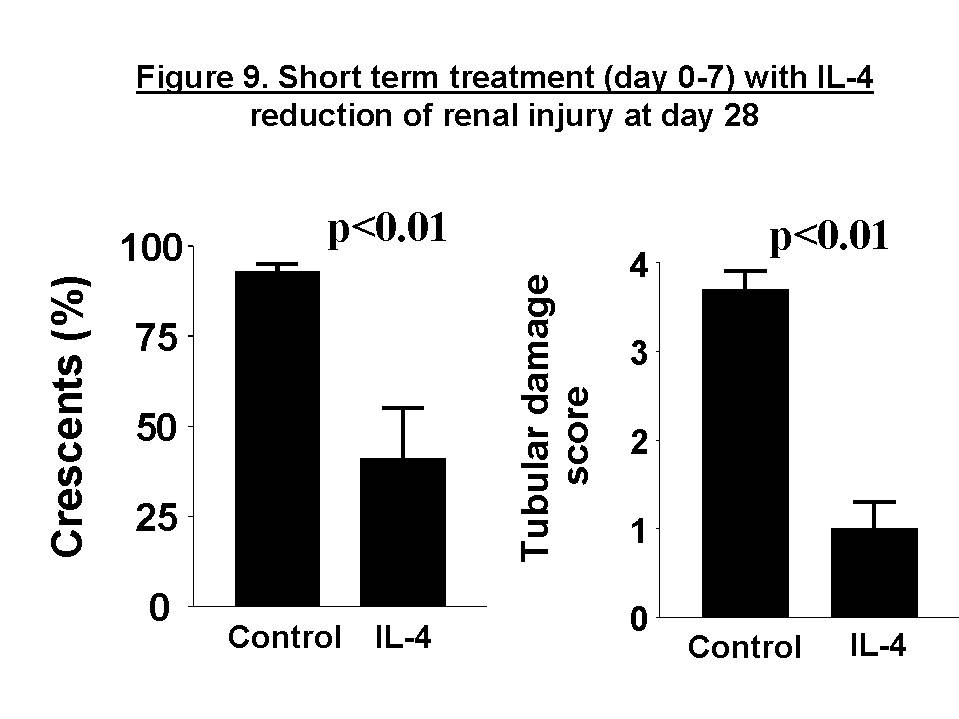 IL-4 is also an important product of type 2 T helper cells (Th2) and has inhibitory effects on the development of Th1 cells and cell mediated immunity. IL-4 knockout mice developed more severe crescentic glomerulonephritis in association with enhanced Th1 response following induction of accelerated nephrotoxic nephritis20. Interleukin-6 The importance of IL-6 in glomerulonephritis is controversial. Overexpression of IL-6 in transgenic mice is known to be associated with massive plasmacytosis in multiple organs and mesangio-proliferative glomerulonephritis. Production of IL-6 is often used as a surrogate marker of acute phase reaction in vivo. Administration of recombinant IL-6 exacerbated glomerulonephritis in (NZB x NZW) F1 mice, but did not induce glomerulonephritis in the parental strain. However, the effect was shown to be related to immune responses rather than a direct pathogenetic role in glomerulonephritis21. However, systemic injection of recombinant human IL-6 has been shown to reduce glomerular synthesis of IL-1 Interleukin-10 IL-10 is very effective in suppressing cytokine production from macrophages. However, its effect in experimental glomerulonephritis is more controversial. Conflicting results have been reported depending on the experimental models used24-25. Interleukin-11 IL-11 can suppress activation in NF- 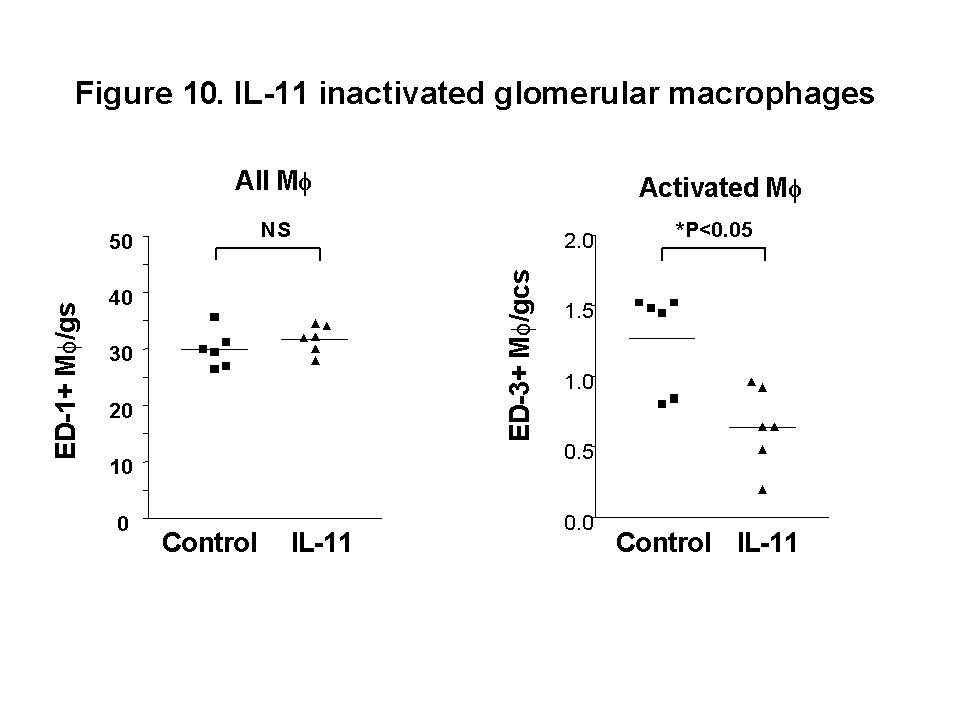 Clinical application At present, there are ongoing clinical trials in the use of monoclonal antibodies or soluble receptor to TNF in patients with renal vasculitis. Recombinant IL-1 receptor antagonist, has been tried in rheumatoid arthritis, and should be considered for use in the treatment of crescentic glomerulonephritis. Further work with anti-inflammatory cytokines and low molecular weight inhibitors of cytokines will provide further therapeutic options. Acknowledgment I would like to express my gratitude to Miss Jennifer Smith for her advice in the preparation of this presentation. References 1. Tam FWK, Pusey CD. The role of T lymphocytes in extracapillary glomerulonephritis. J.Nephrol. 1995;8:305-16. 2. Timoshanko JR, Holdsworth SR, Kitching AR, Tipping PG. IFN-g Production by Intrinsic Renal Cells and Bone Marrow-Derived Cells Is Required for Full Expression of Crescentic Glomerulonephritis in Mice. J Immunol 2002;168(8):4135-41. 3. Timoshanko JR, Kitching AR, Holdsworth SR, Tipping PG. Interleukin-12 from Intrinsic Cells Is an Effector of Renal Injury in Crescentic Glomerulonephritis. J Am Soc Nephrol 2001;12(3):464-71. 4. Garcia GE, Xia Y, Ku G, Johnson RJ, Wilson CB, Feng L. IL-18 translational inhibition restricts IFN-g expression in crescentic glomerulonephritis. Kidney International 2003;64(1):160-9. 5. Kitching AR, Tipping PG, Kurimoto M, Holdsworth SR. IL-18 Has IL-12-Independent Effects in Delayed-Type Hypersensitivity: Studies in Cell-Mediated Crescentic Glomerulonephritis. J Immunol 2000;165(8):4649-57. 6. Timoshanko JR, Sedgwick JD, Holdsworth SR, Tipping PG. Intrinsic Renal Cells Are the Major Source of Tumor Necrosis Factor Contributing to Renal Injury in Murine Crescentic Glomerulonephritis. J Am Soc Nephrol 2003;14(7):1785-93. 7. Tam FWK, Karkar AM, Smith J, Yoshimura T, Steinkasserer A, Kurrle R et al. Differential expression of macrophage inflammatory protein-2 and monocyte chemoattractant protein-1 in experimental glomerulonephritis. Kidney Int. 1996;49:715-21. 8. Wada T, Yokoyama H, Furuichi K, Kobayashi K-I, Harada K, Naruto M et al. Intervention of crescentic glomerulonephritis by antibodies to monocyte chemotactic and activating factor (MCAF/MCP-1). FASEB J. 1996;10:1418-25. 9. Feng L, Chen S, Garcia GE, Xia Y, Siani MA, Botti P et al. Prevention of crescentic glomerulonephritis by immunoneutralization of the fractalkine receptor CX3CR1 rapid communication. Kidney Int 1999;56(2):612-20. 10. Lloyd CM, Minto AW, Dorf ME, Proudfoot A, Wells TNC, Salant DJ et al. RANTES and monocyte chemoattract protein-1 (MCP-1) play an important role in the inflammatory phase of crescentic nephritis, but only MCP-1 is involved in crescent formation and interstitial fibrosis. J.Exp.Med. 1997;185:1371-80. 11. Anders HJ, Frink M, Linde Y, Banas B, Wornle M, Cohen CD et al. CC chemokine ligand 5/RANTES chemokine antagonists aggravate glomerulonephritis despite reduction of glomerular leukocyte infiltration. J Immunol 2003;170(11):5658-66. 12. Tam FWK. Role of selectins in glomerulonephritis. Clin.Exp.Immunol 2002;129(1):1-3. 13. Tam FWK, Smith J, Cashman SJ, Wang Y, Thompson EM, Rees AJ. Glomerular expression of interleukin-1 receptor antagonist and interleukin-1b genes in antibody-mediated glomerulonephritis. Am.J.Pathol. 1994;145:126-36. 14. Lan HY, Nikolic-Paterson DJ, Zarama M, Vannice JL, Atkins RC. Suppression of experimental crescentic glomerulonephritis by interleukin-1 receptor antagonist. Kidney Int. 1993;43:479-85. 15. Tam FWK, Smith J, Karkar AM, Pusey CD, Rees AJ. Interleukin-4 ameliorates experimental glomerulonephritis and upregulates glomerular gene expression of IL-1 decoy receptor. Kidney Int. 1997;52:1224-31. 16. Karkar AM, Smith J, Pusey C.D. Prevention and treatment of experimental crescentic glomerulonephritis by blocking tumour necrosis factor a. Nephrol.Dial.Transplant 2001;16:518-24. 17. Khan, S. B., Bhangal, G., Smith, J., Cook, H. T., and Pusey, C. D. Blockade of TNFa is effective in the treatment of experimental crescentic glomerulonephritis. J Am.Soc.Nephrol 2003. Abstract In Press
18. Tam FWK, Smith J, Agarwal S, Karkar AM, Morel D, Thompson EM et al. Type IV phosphodiesterase inhibitor is effective in prevention and treatment of experimental crescentic glomerulonephritis. Nephron 2000;84(1):58-66. 19. Cook HT, Singh SJ, Wembridge DE, Smith J, Tam FW, Pusey CD. Interleukin-4 ameliorates crescentic glomerulonephritis in Wistar Kyoto rats. Kidney Int 1999;55(4):1319-26. 20. Kitching AR, Tipping PG, Mutch DA, Huang XR, Holdsworth SR. Interleukin-4 deficiency enhances Th1 responses and crescentic glomerulonephritis in mice. Kidney Int. 1998;53(1):112-8. 21. Suematsu S, Matsuda T, Aozasa K, Akira S, Nakano N, Ohno S et al. IgG1 plasmacytosis in interleukin 6 transgenic mice. Proc.Natl.Acad.Sci.U.S.A 1989;86(19):7547-51. 22. Ryffel B, Car BD, Gunn H, Roman D, Hiestand P, Mihatsch MJ. Interleukin-6 exacerbates glomerulonephritis in (NZB x NZW)F1 mice. Am J Pathol 1994;144(5):927-37. 23. Karkar AM, Tam FWK, Proudfoot A, Meager A, Rees AJ. Modulation of antibody-mediated glomerular injury in vivo by interleukin-6. Kidney Int. 1993;44:967-73. 24. Chadban SJ, Tesch GH, Lan HY, Atkins RC, Nikolic-Paterson DJ. Effect of interleukin-10 treatment on crescentic glomerulonephritis in rats. Kidney Int 1997;51(6):1809-17. 25. Huang XR, Kitching AR, Tipping PG, Holdsworth SR. Interleukin-10 inhibits macrophage-induced glomerular injury. J Am Soc Nephrol 2000;11(2):262-9. 26. Lai PC, Cook HT, Smith J, Keith JC, Jr., Pusey CD, Tam FWK. Interleukin-11 attenuates nephrotoxic nephritis in Wistar Kyoto rats. J Am.Soc.Nephrol. 2001;12(11):2310-20. |
 therapy in patients with renal vasculitis.
therapy in patients with renal vasculitis. and IL-12 are important in infiltration of macrophages and T cells and crescentic formation2, 3.
and IL-12 are important in infiltration of macrophages and T cells and crescentic formation2, 3. 
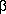 , RANTES
, RANTES
 B which is a critical transcription factor in the development of glomerular inflammation. Administration of IL-11 was shown to reduce the severity of nephrotoxic nephritis in WKY rats. Both mesangial cells and macrophages express IL-11 receptors26. When a low dose of IL-11 was used, it was demonstrated that glomerular macrophage activation and injury were reduced without affecting the total number of macrophages (Figure 10).
B which is a critical transcription factor in the development of glomerular inflammation. Administration of IL-11 was shown to reduce the severity of nephrotoxic nephritis in WKY rats. Both mesangial cells and macrophages express IL-11 receptors26. When a low dose of IL-11 was used, it was demonstrated that glomerular macrophage activation and injury were reduced without affecting the total number of macrophages (Figure 10).