El síndrome de DiGeorge (SDG) es un trastorno citogenético debido a una deleción cromosómica de 22q11, que da lugar a un defecto del desarrollo de las bolsas faríngeas 3ª y 4ª (1). Como consecuencia, se producen hipoplasia/aplasia del timo (con inmunodeficiencia de células T en el 19 a 25 % de los pacientes) y de paratiroides (con hipocalcemia); anomalías cardíacas congénitas, principalmente del tracto de salida ventricular, y rasgos dismórficos característicos. Actualmente se incluye al SDG en el llamado «síndrome de deleción del cromosoma 22q11» junto al síndrome velocardiofacial, que se acompaña con mayor frecuencia que el SDG de alteraciones dismórficas y cardíacas, y en el que es menos frecuente la inmunodeficiencia.
Los pacientes con síndromes de inmunodeficiencia congénita presentan un riesgo aumentado de desarrollar neoplasias malignas (2), en particular linfomas no Hodgkin (LNH); sin embargo, ésto no es así en lo que se refiere al SDG. El caso que recoje esta presentación es único debido a que no existe en la literatura médica ningún caso descrito de LNH en pacientes con SDG.
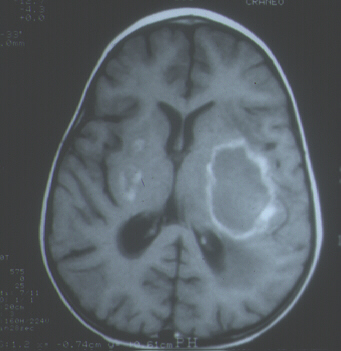
Paciente de sexo femenino, de 2 años de edad, sin dismorfias faciales, paladar hendido ni anomalías cardíacas, en la que a los 10 meses de vida se estableció el diagnóstico de SDG debido a la detección de hipocalcemia (calcio sérico, 1,5 mmol/l) por hipoparatiroidismo (paratohormona sérica, 6,7 pg/ml) y déficit de células T. Entre los 21 y 23 meses de edad presentó una masa hiperecogénica hepática y un cuadro de hemiparesia espástica derecha progresiva, detectándose una gran lesión ocupante de espacio en hemisferio cerebral izquierdo y dos pequeñas lesiones en ganglios basales derechos mediante tomografía computadorizada y resonancia magnética (Fig. 1). Se realizó una biopsia cerebral estereotáxica. La niña falleció un mes después y se efectuó el estudio autópsico. Sobre la biopsia cerebral y los tejidos obtenidos en la autopsia se realizaron técnicas de macroscopia y microscopia convencionales, así como de inmunohistoquímica para fenotipificación de las células tumorales e hibridación in situ para demostración del ARN del virus de Epstein-Barr (VEB).
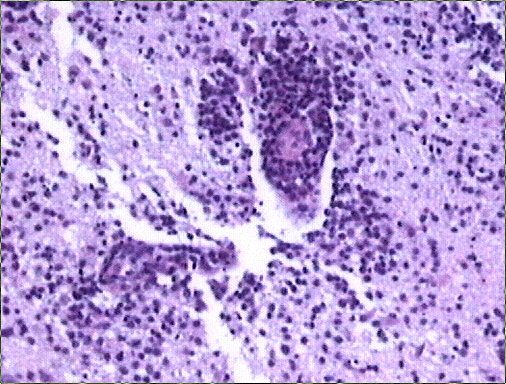
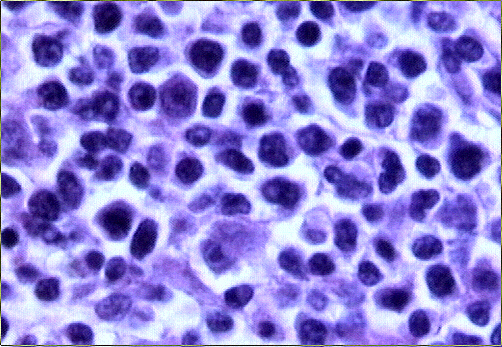
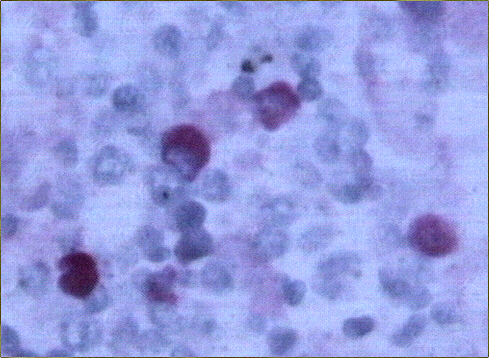
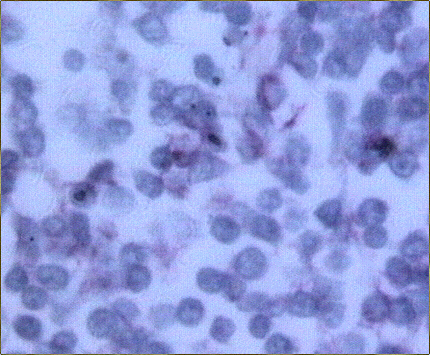
El estudio histológico de la biopsia cerebral estereotáxica reveló un parénquima neural con amplias áreas de necrosis tumoral en relación con una proliferación de células de hábito linfoplasmocitario, monomorfas, de distribución predominantemente perivascular (Figs. 2-4) y con suficiente atipia citológica como para establecer el diagnóstico de presunción de linfoma no Hodgkin de alto grado de malignidad.
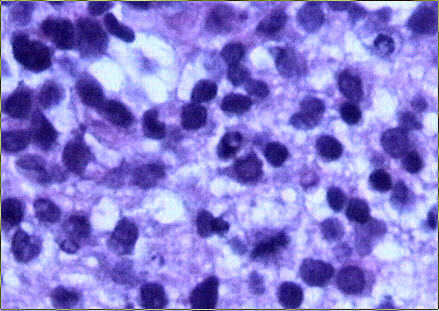
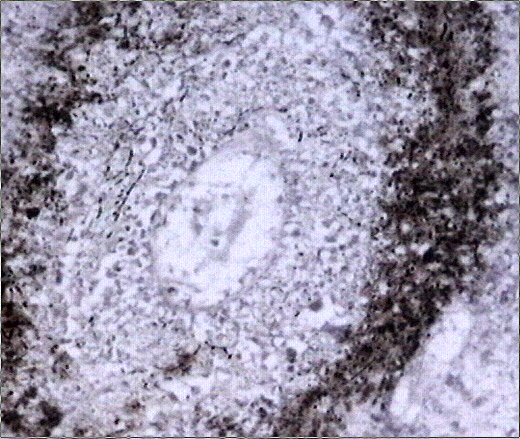
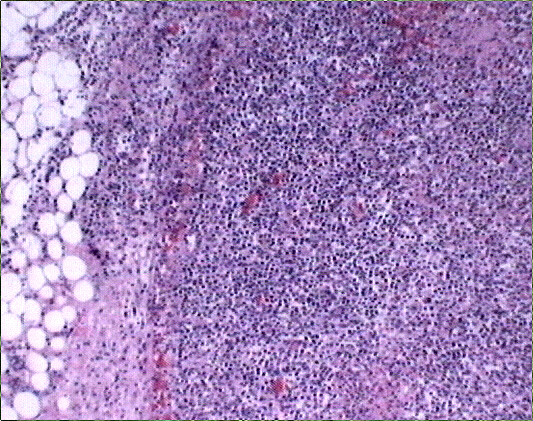
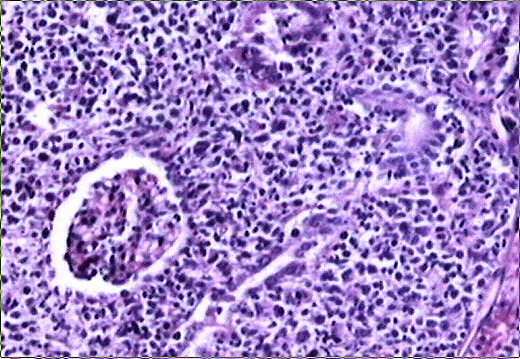
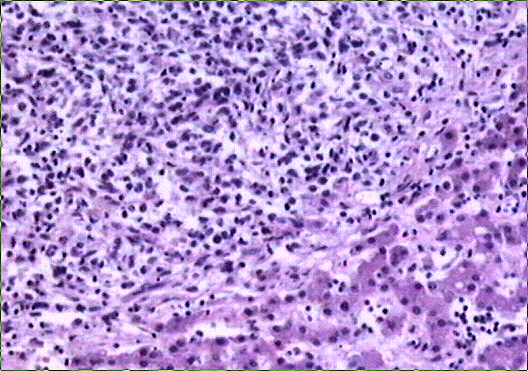
El estudio macroscópico post-mortem demostró ausencia completa de timo y paratiroides, ausencia de defectos cardiovasculares y presencia de lesiones ocupantes de espacio en ganglios linfáticos mediastínicos, hígado, riñones y cerebro. Estas lesiones correspondían microscópicamente a la infiltración de dichos órganos por un linfoma no Hodgkin de alto grado, de tipo linfoblástico. Las células neoplásicas crecían en sábana y mostraban núcleos frecuentemente convolucionados, con cromatina finamente granular y nucléolo visible. La actividad mitósica era frecuente. En los ganglios mediastínicos, el linfoma borraba completamente la arquitectura ganglionar y crecía de forma difusa, infiltrando el tejido adiposo periganglionar (Fig. 5), con unas características citológicaas idénticas a las observadas en la biopsia estereotáxica (Fig. 6). En ambos riñones, el patrón de infiltración era predominantemente intersticial, rodeando a los glomérulos y túbulos nativos y asociándose a amplias áreas de necrosis (Fig. 7). En hígado formaba grandes masas asociadas a necrosis extensa que reemplazaban el tejido hepático normal (Fig. 8). Por último, se objetivó una infiltración masiva de ambos hemisferios cerebrales por el linfoma, con extensa necrosis, reacción histiocitaria intensa y un patrón característico de crecimiento perivascular, similar a lo ya expuesto. El estudio inmunohistoquímico reveló positividad de las células neoplásicas para marcadores linfoides B (CD 20) (Fig. 9) y negatividad para marcadores T (CD 3, CD45RO) y cadenas ligeras kappa y lambda.
La técnica de hibridación in situ mostró la expresión masiva de transcripciones EBER (ARN codificado por el VEB) en las células tumorales. El diagnóstico final fue el de linfoma de alto grado linfoblástico tipo B.
La aparición de un LNH en pacientes con SDG es excepcional. En otras inmunodeficiencias congénitas se han descrito, sin embargo, riesgos elevados de desarrollo de LNH respecto al total de tumores malignos: 46 % en pacientes con ataxia-telangiectasia, 48,8% en la inmunodeficiencia variable común, 75,6 % en el síndrome de Wiskott-Aldrich y 73,8 % en la inmunodeficiencia combinada severa (2). Una posible explicación del escaso desarrollo de LNH en el SDG, aunque todavía sin dilucidar, podría ser la baja frecuencia con que este síndrome ocurre de forma completa, es decir, asociando inmunodeficiencia celular severa (19 a 25 % de los casos); la forma más frecuente es la parcial, con preservación del número y función de las células T (3).
Los linfomas en pacientes con inmunodeficiencias se presentan con frecuente extensión extranodal, particularmente afectando al tracto gastrointestinal y al sistema nervioso central. En el presente caso, la afectación extraganglionar también es extensa (4).
En la patogenia de los LNH desarrollados en pacientes con inmunodeficiencia se ha implicado al virus de Epstein-Barr (VEB). La primoinfección por VEB suele ocurrir en la infancia o adolescencia, generalmente en forma de enfermedad autolimitada en la que una pequeña población de linfocitos B queda infectada de forma latente, persistiendo durante toda la vida del individuo. Aunque estos linfocitos pueden crecer de forma acelerada en presencia de un sistema inmune deficiente, el VEB no causa la transformación maligna de las células. Se ha postulado que el crecimiento celular inducido por el VEB aumenta la vulnerabilidad de las células a adquirir mutaciones, bien por un segundo agente o por recombinación genética (5). El sistema inmune del individuo sano, sobre todo la inmunidad celular, suprime el crecimiento de las células infectadas por VEB, pero éstas pueden desarrollar un crecimiento neoplásico en caso de inmunodeficiencia. El VEB también se ha aislado en casos de enfermedad de Hodgkin, en el linfoma de Burkitt (con mayor frecuencia en la variedad endémica), en los LNH asociados a la infección por el virus de la inmunodeficiencia humana (VIH) y a los procesos linfoproliferativos desarrollados post-trasplante. Por tanto, en nuestro caso (4), la infección por el VEB en un paciente con inmunodeficiencia celular puede haber favorecido la aparición de un LNH.
Síndrome de DiGeorge - Virus de Epstein-Barr - Linfoma cerebral - Inmunodeficiencia - Timo.
|
|