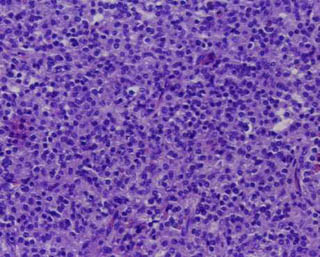
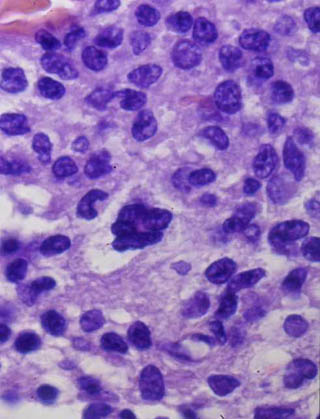
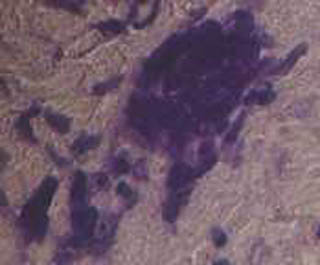
Imagen 6. Nótese la negatividad de la S-100. (100x).
El xantogranuloma juvenil (XGJ), -nevoxantoendotelioma-, es una patología histiocítica benigna típica de la infancia. Clínicamente se caracteriza por la aparición de una o más lesiones nodulares cutáneas cuya tendencia natural es la regresión espontánea. Sólo de manera excepcional existe afectación sistémica que puede acompañarse o no de lesiones cutáneas. Es por ello que son pocos los casos comunicados de XGJ sistémico y siempre de manera aislada. Entre los órganos extracutáneos afectados se encuentran: ojos, cavidad oral, pulmón, pleura, pericardio, hígado, riñón, testículo, músculo esquelético, ganglios linfáticos... La afectación del sistema nervioso central (SNC) también se ha descrito (9 casos sintomáticos) y en sólo 2 de ellos hay comprobación histopatológica. Presentamos un caso de XGJ cutáneo que desarrolló lesiones sistémicas en pulmón y SNC. Éstas últimas provocaron clínica por lo que fue necesario tratamiento quirúrgico. Mostramos la histología encontrada así como una revisión de la situación actual de esta entidad.
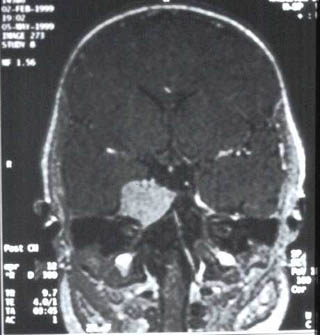
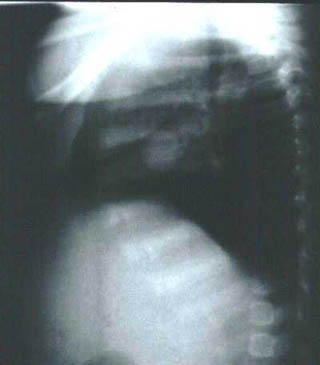
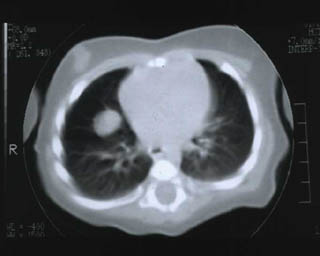
Lactante (sexo femenino) de 3 meses de edad sin antecedentes obstétricos de interés que presenta desde el nacimiento dos nódulos cutáneos amarillo-rosados, uno en cuadrante inferior izquierdo de abdomen (3x2 cm) y otro preauricular izquierdo (3 mm). Muestra otro subcutáneo de 0,5 cm en región subcostal ( línea axilar anterior). El nódulo abdominal había aumentado progresivamente de tamaño por lo que se biopsió (en otro centro) siendo informado como xantogranuloma juvenil. Ingresa en nuestro hospital por un cuadro de paresia del VI par craneal derecho. El estudio de extensión ( Rx de tórax, TAC y RM) mostró dos lesiones cerebrales, ambas en relación directa con la duramadre: una pequeña, hipercaptante, en base meníngea frontal izquierda y otra en seno cavernoso derecho (2 cm) (imagen 1). Existían además dos nódulos pulmonares (1,5 y 1,2 cm respectivamente) en lóbulo medio derecho y lóbulo superior izquierdo que no producían sintomatología (imagen 2 y 3). El estudio oftalmológico (fondo de ojo) y la analítica (incluyendo perfil lipídico) fueron normales. Se decidió tratamiento quirúrgico de la mayor de las lesiones cerebrales y mediante abordaje pterional-subtemporal se extirpó por completo. El postoperatorio transcurrió sin incidencias y la niña fue dada de alta.
El estudio histológico de la lesión tumoral cerebral (procesados según técnica rutinaria en nuestro departamento: fijación en formaldehído al 10%, inclusión en parafina y tinción con hematoxilina-eosina) mostró una proliferación celular monomorfa, de tamaño intermedio, núcleo redondeado y citoplasma mal delimitado (imagen 4). Focalmente se reconocían células gigantes multinucleadas de hábito histiocitario similares a las de Touton (imagen 5). No se observaron fenómenos de emperipolesis, eritrofagocitosis, necrosis o fibrosis. Las técnicas histoquímicas para demostración de agentes infecciosos fueron negativas. El estudio inmunohistoquímico realizado sobre tejido fijado en formol e incluído en parafina mostró que las células proliferantes eran negativas para S-100 (imagen 6), CD-1a, HMB-45, CD-68, CD-20, Kappa y Lambda. Sólo había positividad focal para MAC-387 y para aislados linfocitos CD 3 de carácter reactivo. En base a la histología y a una inmunohistoquímica compatible se diagnosticó de xantogranuloma juvenil cerebral.
El XGJ es una entidad que se incluye dentro de las histiocitosis(1) o transtorno caracterizado por proliferación de células del sistema mononuclear fagocítico. Se piensa que los histiocitos proceden de células madre de la médula ósea. De estos histiocitos derivan posteriormente los monocitos-macrófagos, (cuya función es la fagocitosis) y las células de Langerhans (que actuan como presentadoras de antígenos a los linfocitos T CD4 para así iniciar la respuesta inmune). Según Cline (2) el XGJ, el xantoma diseminatum, el reticulohistiocitoma multicéntrico y la histiocitosis nodular progresiva se originarían a partir de proliferaciones de la serie monocitaria-macrofágica y todos ellos representarían la misma lesión en diferentes estadios evolutivos (3). Por el contrario, la histiocitosis X se originaría a partir de proliferación de las células de Langerhans y este cuadro debe distinguirse del anterior por su distinto pronóstico y tratamiento.
El XGJ afecta pricipalmente a lactantes y niños menores de 10 años1, con incidencia algo mayor en varones (4:1) (4,5). Se han descrito casos en adultos (incidencia del 17,5 % o del 23,4 % según series(4,6)). La clínica consiste en papulo-nódulos cutáneos amarillentos-rojizos constituídos por un infiltrado transdérmico o en dermis reticular de histiocitos mononucleados, formas multinucleadas tipo Touton en cantidad variable y en ocasiones eosinófilos (planteándose el diagnóstico diferencial con la histiocitosis de Langerhans. La histología suele ser diagnóstica pero en casos dudosos las células del XGJ son negativas para la S-100 y no contienen granos de Birbeck). Las lesiones siguen un curso evolutivo benigno con estabilización o regresión espontánea en la mayoría de los casos (7). Basicamente existen dos formas clínicas: cutánea y sistémica. La cutánea fue descrita en 1905 por Adamson (8) e inicialmente llamada xantoma congénito múltiple. Posteriormente McDonough (9) propuso un origen endotelial, de ahí la denominación de nevoxantoendotelioma. Serán Helwig y Hackney (10) quienes determinen su origen fibrohistiocitario acuñando el nombre de xantogranuloma. La clínica es la descrita previamente. Se ha asociado a Leucemia Mieloide Crónica Juvenil (11-13), a neurofibromatosis tipo I (12-15), a diabetes mellitus insulin dependiente (16), a urticaria pigmentosa (17,18), prurito acuagénico (19) y posible infección por CMV (20). No se ha encontrado asociación con hiperlipidemia 1ª 4. La afectación sistémica (5-10%)1 debe considerarse siempre que existan lesiones cutáneas, requiriendo estudios de extensión sólo si hay sintomatología (en nuestro caso se realizó al iniciarse la paresia del VI PC). En el estudio realizado por Freyer (1) en el que analiza 36 casos recogidos en la literatura, se observa que en ocasiones existe afectación sistémica sin afectación cutánea y que si coexisten ambas, la cutánea suele preceder a la sistémica. Además, son las lesiones cutáneas macronodulares las que se asocian a formas sistémicas (7,21). Las localizaciones extracutáneas más frecuentes son ojo (22) ,(por ello es obligatorio un estudio oftalmológico en estos niños), tejido celular subcutáneo, hígado, bazo, pulmón y SNC. También se han descrito en músculo, periostio, glándula salival y mucosas (24) . La resección quirúrgica cuando es posible es curativa. En SNC la clínica que producen se debe al afecto de masa: diplopia, epilepsia, ataxia, neuralgias, aumento de presión intracraneal, retraso en el desarrollo...(24-26). Suelen ser lesiones múltiples afectando cerebro, cerebelo, región periventricular, leptomeninges, médula espinal y cavidad trigeminal. Es necesario hacer diagnóstico diferencial con otras lesiones histiocíticas del SNC como el xantoma diseminatum (realmente variante de la misma enfermedad), la histiocitosis sinusal con linfadenopatía masiva (suele haber afectación ganglionar con fiebre y leucocitosis, así como emperipolesis), con el xantogranuloma del plexo coroideo (hallazgo casual de autopsias, con nidos de histiocitos, cristales de colesterol, células gigantes e inflamación crónica), histiocitosis X, quistes epiteliales y sobre todo es importante descartar el linfoma (estudio inmunohistoquímico) pues pueden tener aspecto histiocítico (26). En los casos con afectación del SNC la cirugía curativa no suele ser posible y se han intentado otras medidas terapeúticas que incluyen radioterapia y poliquimioterapia (similar a la usada en la histiocitosis de Langerhans) (27) con distintos resultados -dos casos fallecieron por la afectación cerebral (1). En nuestra paciente la cirugía fue suficiente. El uso de estos tratamientos está sujeto a controversia por los efectos secundarios de la quimio y radioterapia en niños, en los que además estamos tratando unas lesiones cuya tendencia espontánea frecuentemente es la regresión. Se necesitarían estudios más amplios para optimizar el tratamiento adecuado en los pacientes con clínica en los que no es posible una cirugía.
1. Freyer DR, Kennedy R, Brostrom B C, et al. Juvenile xanthogranuloma: forms of systemic disease and their clinical implications. The Journal of Pediatrics 1996;129: 227-237.
2. Cline MJ. Histiocytes and histiocytosis. Blood 1994;84:2840-53.
3. Marrogi AJ, Dehner LP et al. Benign cutaneous histiocytic tumors in childhood and adolescence excluding Langerhans cells proliferations. Am J Dermatopathol 1992; 14:8-18.
4. Cohen B, Hood A. Xanthogranuloma: report on clinical and histologic findings in 64 patients. Pediatr Dermatolo 1990; 6: 262-266.
5. Tahan SR, Pastel-Levy C et al. Juvenile Xanthogranuloma: clinical and pathologic characterization. Arch Pathol Lab Med 1989;113: 1057-61.
6. Sonoda T, Hashimoto H, Enjoi H. Juvenile Xanthogranuloma. Clinicopathologic analysis and immunohistochemical study of 57 patients. Cancer 1985; 56: 2280-2286.
7. Gianotti F, Caputo R. Histiocytic syndromes: a review. J Am Acad Dermatol. 1985; 13: 383-404.
8. Adamson HG. Society intelligence: The dermatological society of London. Br J Dermatol 1905;17:222.
9. McDonough JFR: a contribution to our Knowledge of the nevoxanthoendotelioma. Br J Dermatol 1912;24:85-99.
10. Helwig EB, Hacney VC: juvenile xanthogranuloma (nevoxanthoendotelioma). Am J pathol 1954;30.625-626.
11. Cooper PH, Frierson HF et al. Associaton of juvenile xanthogranuloma with juvenile myeloid leukemia. Arch Dermatol 1984; 120:371-5.
12. Ludwig J, Wester S et al. Evidence of neurofibramatoses and chronic myelogenous leukemia in a liver biopsy specimen (letter). J Clin Gastroenterol 1993; 16: 265-7.
13. Morier P, Merot Y et al. Juvenile chronic granuloctic leukemia, juvenile xanthogranulomas, and neurofibromatosis. J Am Acad Dermatol 1990; 22:962-5.
14. Sillevis-Smith JH. Juvenile Xanthogranuloma in neurofibromatosis tipo I. Br J Dermatol 1991;125:390.
15. Ackerman CD, Cohen BA. . Juvenile Xanthogranuloma and neurofibramatosis. Pediatr Dermatol 1991;8:339-40.
16. Ginsberg-Fellner F, Fellner MJ. Juvenile Xanthogranuloma in a child with insulin-dependant diabetes mellitus. Int J Dermatol 1982;21:36-9.
17. De Villez RL, Limmer BL. Juvenile Xanthogranuloma and Urticaria Pigmentosa. Arch Dermatol 1975; 11:365-6
18. Nagayo K, Sakai M, Minuzo N. Juvenile Xanthogranuloma with Darier's sign. J Dermatol 1983; 10:283-5.
19. Handfield-Jones SE, Hills RJ, et al. Aquagenic pruritus associated with juvenile xanthogranuloma. Clin Exper Dermatol 1993;18:36-9.
20. Balfour HH, Speicher CE, et al. Juvenile Xanthogranuloma associated with cytomegalovirus infection. AM J Med 1971;50:380-4.
21. Botella-Estrada R, Sanmartín O, Grau M, et al. Juvenile Xanthogranuloma with Central Nervous System involvement. Pediatric Dermatology 1993; 10, 1: 64-68.
22. Roper SS, Spraker MK. Cutaneous histiocytosis syndromes. Pediatr Dermatol 1985; 3:19-30.
23. Wolff HM, Vigl E, Braun-Falco O. Juveniles xanthogranulom und organ manifestationen. Hautarzt 1975; 26:268-272.
24. Chu AC, Wells RS, MacDonald DM,. Juvenile Xanthogranuloma with recurrent subdural effusions. Br J Dermatol 1981; 105: 97-101.
25. Flach DB, Winkelmann RK. Juvenile Xanthogranuloma with central nervous system lesions . J Am Acad Dermatol 1986; 14: 405-411.
26. Paulus W, Kirchner T, Ott MM et al. Histiocytic tumor of Meckel's cave. Am J Surg Pathol 1992; 16: 76-83.
27. Schultz KD, Petronio J et al. Solitary intracerebal juvenile Xanthogranuloma. Pediatr Neurosurg 1997; 26:315-321.
|
Imagen 6. Nótese la negatividad de la S-100. (100x). |