

ASPECTOS ANATOMOPATOLÓGICOS EN TRES CASOS DE SÍNDROME DE VON HIPPEL-LINDAU
AUTORES: Dr. Eduardo Brandán Recalde; Dra. Mónica Matsuzaki;
Dra. María E. Andurno.
1º Cátedra de Patología.
Hospital Nacional de Clínicas.
Universidad Nacional de Córdoba.
República Argentina.
INTRODUCCIÓN
El síndrome de von Hippel-Lindau (VHL) es un trastorno genético poco común descripto a comienzos del año 1900 por von Hippel y posteriormente Lindau (1) como una alteración genética autosómica dominante. Se presenta con varias lesiones. Entre las más frecuentes, se encuentran: hemangioblastomas en retina, cerebelo y médula espinal, (2) feocromocitoma, Cistoadenoma Seroso Microquístico (CSM), especialmente de localización pancreática, quistes renales y carcinomas de células renales. (3) Estos últimos se desarrollan en un 38% a 55%. A menudo son bilaterales, multicéntricos y, en general, se expresan en pacientes más jóvenes que los casos esporádicos. (4) En ocasiones, se han descripto sólo carcinomas de células renales periféricos y CSM pancreáticos. También han sido mencionadas lesiones angiomatosas en hígado, riñón, páncreas, pulmón, piel y epidídimo (1,3). Por otro lado las comunicaciones de presentaciones de CSM en mesosalpinx y ligamento redondo expresan localizaciones diferentes a las más comunes en el páncreas. Comunicamos tres casos, (caso Nº 1) mujer joven, con asociación de lesión quística pancreática, antecedentes familiares de (caso Nº 2) hemangioblastoma cerebelar en una hermana (joven), operada en el Hospital Privado de Córdoba (R. A.) en el mes de septiembre de 1998 (madre fallecida por probable tumor de SNC, padre fallecido por probable tumor renal), quistes bilaterales de riñón, carcinomas renales periféricos multicéntricos y CSM en apéndice cecal, y (caso Nº 3) cistoadenoma seroso microquístico y carcinoma renal periférico en una paciente adulta, con antecedentes familiares asociados al síndrome.
PRESENTACIÓN DE LOS CASOS
CASO Nº 1
Mujer de 22 años que en el mes de octubre de 1998 consultó en el Hospital Nacional de Clínicas (Córdoba- R. A.) por un cuadro agudo de fosa ilíaca derecha. Se operó de urgencia. Se encontró una apendicitis aguda supurada, junto a una manifestación tumoral, (Macrofotografía1) que en ausencia de datos clínicos, se sospechó inicialmente de un mesotelioma apendicular concomitante versus la metástasis de un adenocarcinoma túbulopapilar de probable origen renal. (Microfotografía1) Una ecografía abdominopelviana demostraba múltiples imágenes quísticas en páncreas y riñones, un tumor en el polo superior del riñón derecho y microquistes corticales en el izquierdo. (Fotografía de T.A.C.1) (Fotografía de T.A.C.2) Una Tomografía Axial Computarizada posterior, reafirmó los hallazgos iniciales y se complementó la exploración con una RMN, la que evidenció un incremento de la vascularización en varios nódulos del riñón derecho. En la pieza quirúrgica se observaron cuatro nódulos carcinomatosos, multicéntricos, periféricos, y varios quistes con contenido hemático. También se detectaron microangiomas múltiples. (Macrofotografía2)
Las secciones histológicas demostró, en los nódulos de aspecto neoplásico, una proliferación de células de estirpe epitelial conformando estructuras túbulo-papilares tapizadas por células con núcleos pequeños y abundante citoplasma claro. (Microfotografía2)
Observamos, además, la presencia de estructuras quísticas, tapizadas por una monocapa de células cúbicas, con material eosinófilo amorfo en su interior. (Microfotografía3)
Resultaba llamativo en diferentes áreas, la presencia de una miríada de microangiomas. (Microfotografía4)
La revisión del material de la pieza quirúrgica del apéndice cecal, nos permitió reinterpretar la lesión como un Cistoadenoma Seroso Microquístico de localización poco habitual.
CASO Nº 2
Mujer de 21 años (hermana de la paciente del caso Nº 1) que en el mes de septiembre de 1999, consultó por cefaleas, vómitos y vértigo. Por RMN se demostró un tumor en fosa posterior de 4x3x3cm que desplazaba al IV ventrículo. Resultó un hemangioblastoma de cerebelo. (Microfotografía5) (Microfotografía6) Una TAC abdominal no demostró lesiones pancreáticas ni renales.
CASO Nº 3
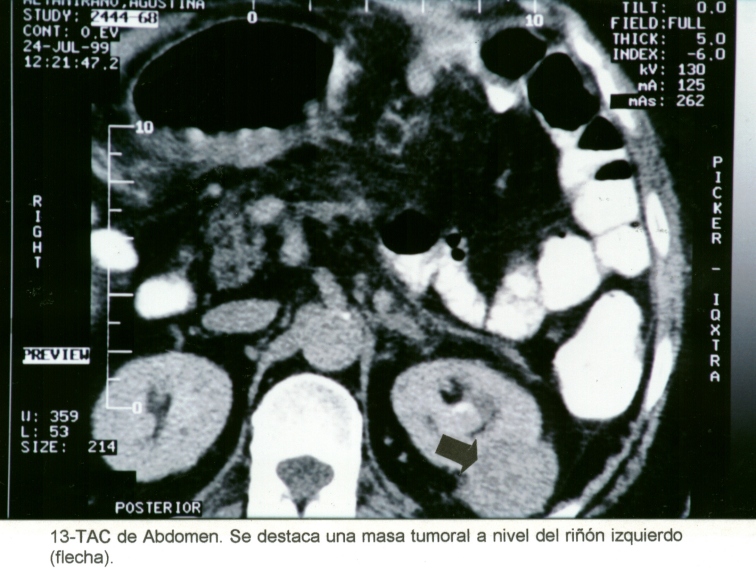
Mujer de 76 años, con antecedentes de hipertensión arterial, operada de un adenocarcinoma de mama en marzo de 1998. En el mes de abril de 1999, se diagnosticó un tumor quístico pancreático, (Macrofotografía3) y un tumor periférico en el riñón izquierdo. (Macrofotografía4) (Fotografía de T.A.C.3) Se practicó primero una pancreatectomía parcial, con un tumor perteneciente a un Cistoadenoma Seroso Microquístico (Microfotografía7) y en el mes de agosto de 1999 se extirpó el riñón izquierdo, con un adenocarcinoma periférico de células claras. (Microfotografía8) Dentro de los antecedentes familiares, con dificultad, se pudo recabar: Madre fallecida por tumor pancreático, padre fallecido por HTA, dos hermanos vivos hipertensos y una hija operada de un tumor cerebral sin precisión, por parte de la paciente, del diagnóstico anatomopatológico realizado en otra institución.
DISCUSIÓN
El síndrome de von Hippel-Lindau conviene investigarlo, toda vez que se detecte en cualquier localización un adenoma seroso microquístico. Es conveniente prestar atención a una metodología de pesquisa cada vez que se lo sospecha, con una detallada historia clínica del paciente y de su familia, un prolijo examen neurológico y oftalmológico, control de la presión arterial, investigación de catecolaminas en orina de 24 horas, ecografía de riñones, páncreas, glándula adrenal y epidídimo. Es adecuado, también, efectuar TAC y RMN de abdomen y cerebro en aquellos pacientes con hallazgos patológicos. (3) Debe tenerse en cuenta, también, los aspectos genéticos de este síndrome que se transmite en forma autosómica dominante con una penetración variable. El gen anormal asociado con este síndrome se localiza en la porción distal del cromosoma 3p; este gen es un oncogen recesivo. La ausencia del gen supresor tumoral conduce al desarrollo de tumores característicos de esta enfermedad. (5) Las personas afectadas deben ser prevenidas en razón que su progenie puede expresar alguna manifestación con el paso del tiempo de esta enfermedad. De nuestros casos, resaltamos el hallazgo, en una localización poco frecuente de cistoadenoma seroso microquístico en apéndice cecal, en el curso de una inflamación aguda, y la presentación del síndrome de von Hippel- Lindau en una mujer de 76 años, con antecedentes familiares asociados.
CASO Nº 1
 Macrofotografía1
Macrofotografía1
 Macrofotografía2
Macrofotografía2
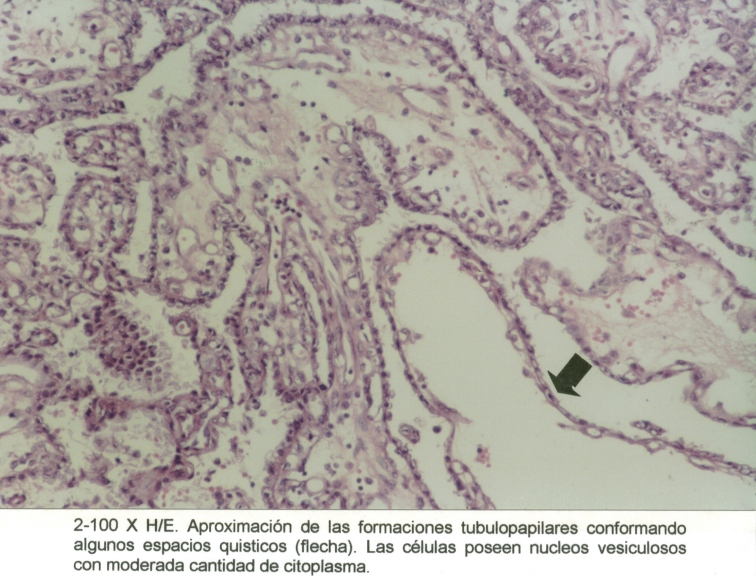 Microfotografía1
Microfotografía1
 Microfotografía2
Microfotografía2
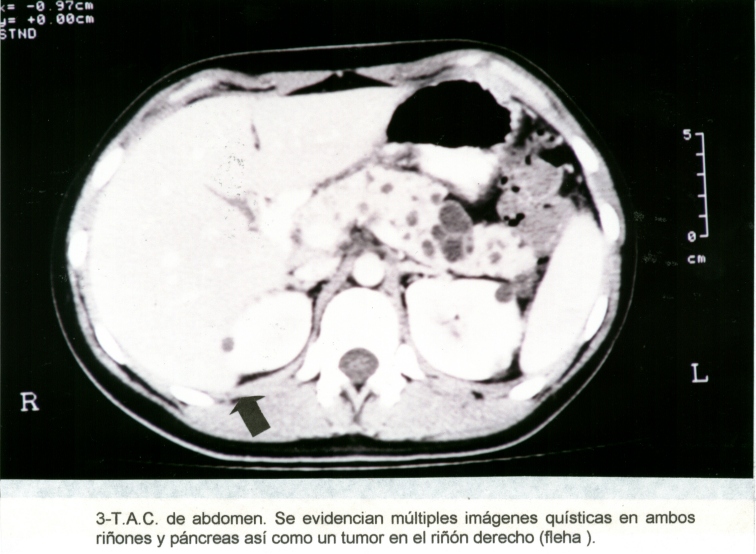 Fotografía de T.A.C.1
Fotografía de T.A.C.1
 Fotografía de
T.A.C.2
Fotografía de
T.A.C.2
CASO Nº 2
 Microfotografía5
Microfotografía5
 Microfotografía6
Microfotografía6
CASO Nº 3
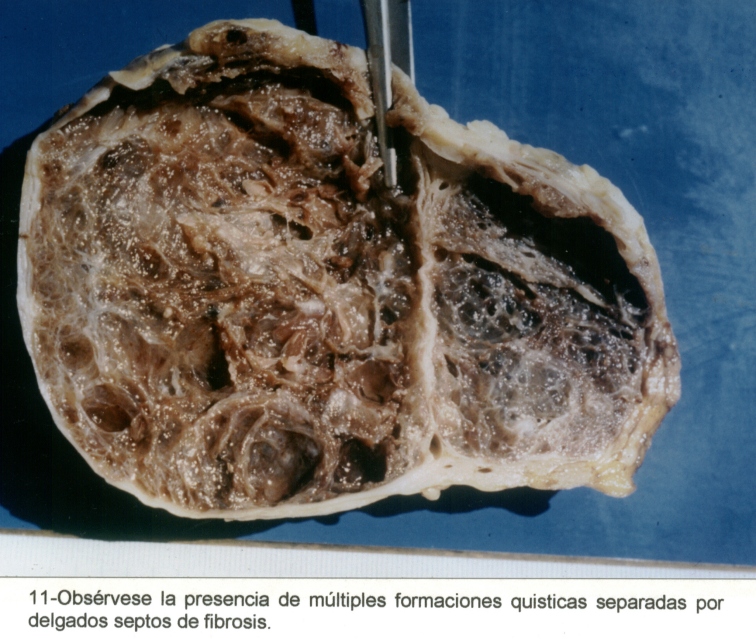 Macrofotografía3
Macrofotografía3
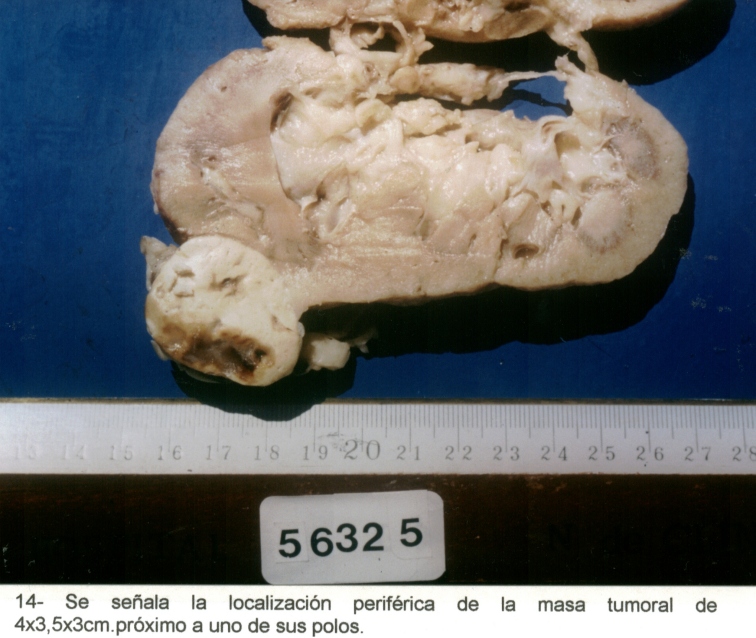 Macrofotografía4
Macrofotografía4
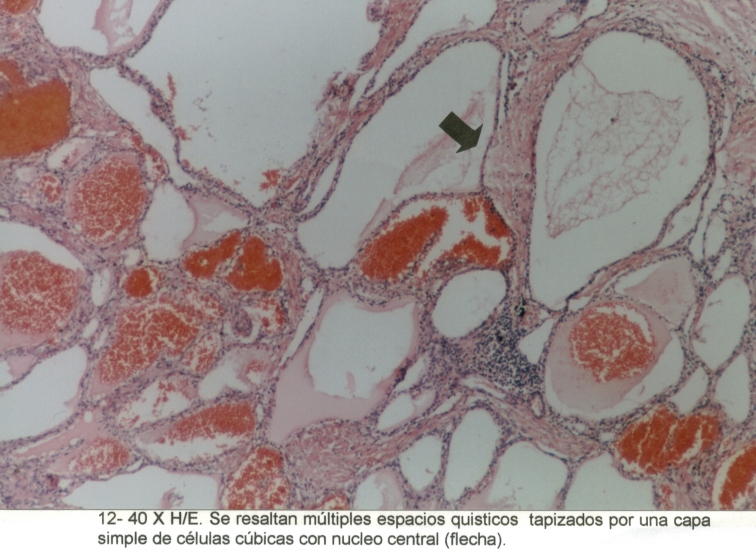 Microfotografía7
Microfotografía7
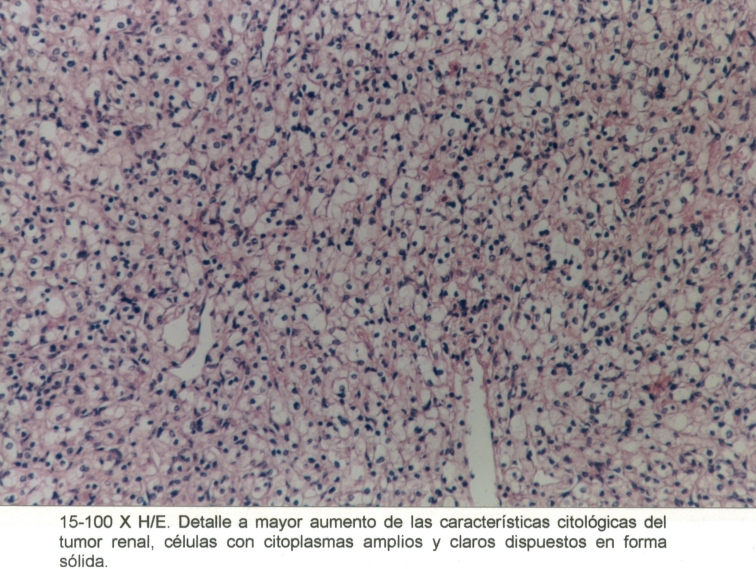 Microfotografía8
Microfotografía8
BIBLIOGRAFÍA
Bell, D.A.; Woodruff, J.M.; Scully, R.E.: Ependymoma of the broad ligament. A report of two cases. Am. J. Surg. Pathol. 8: 203-209. 1984.
Malek, R. S.; Omess, P. J.; Benson, R. C.; Zincke, H.: Renal Cell Carcinoma in von Hippel- Lindau Syndrome. Am. J. Med. Vol. 82: 236-238. 1987
Gersell, D.J.: Papillary cystadenoma of the mesosalpinx in Von Hippel Lindau disease. Am.J. Surg. Pathol. 12: 145-149. 1988.
Solomon, D.; Schwartz, A.: Renal Pathology in von Hippel- Lindau Disease. Pathol. Vol. 19 Nº 9: 1072-1079. 1988.
Alpert, L. C.; Truong, L. D.; Bossart, M. I.; Spjut, H.: Microcystic Adenoma (Serous Cystadenoma) of the pancreas. A study of 14 Cases with Immunohistochemical and Electro-Microscopic Correlation. Am. J. Surg. Pathol. 12(4): 251-263. 1988
Funk. K. C.; Heiben, J. P.: Papillary cystadenoma of the broad ligament in Von Hippel Lindau disease. Am. J. Radiol. 153: 527-528. 1989.
Korn, W. T.; Schattler, S. C.; Disciullo, A. J.; Scully, R. E.: Papillary cystadenoma of the broad ligament in Von Hippel Lindau disease. Am. J. Obstet. Gynecol. 163: 596-598. 1990.
Neumann, H. P.; Dinkel, E. et al.: Pancreatic lesion in the von Hippel-Lindau Syndrome. Gastroenterol. 101: 465-471. 1991.
Richmond, B. K.; Schmidt J. H. III: Congenital cystic supratentorial hemangioblastoma. Case report. J.Neurosurg 82:113-115. 1995.
Sternberg, S.: Diagnostic Surgical Pathology. Vol 2 Third Edition. 1999.
RESEÑA HISTÓRICA
El Hospital Nacional de Clínicas, es el Hospital Escuela de la Facultad de Ciencias Médicas de Córdoba. Fue inaugurado el 24 de mayo de 1913, bajo la dirección del Prof. Dr. Pedro Vella, catedrático titular de Clínica Quirúrgica. La figura señera del Hospital, padre de ilustres médicos que continúan enriqueciendo al país con su saber, fue reconocida por el Poder Ejecutivo Nacional, quién por Decreto Nro 1472/96, lo declaró Monumento Histórico, para continuar con el liderazgo de su accionar entrelazando históricamente el presente con el pasado.

Material digitalizado y compaginado en laboratorios de

Roberto Alejandro Cabanillas Acerbi

{kind=link}
{kind=link}