RESUMEN
Se evaluaron los expedientes clínicos de dos pacientes atendidos
en el Servicio de Cabeza y Cuello del Instituto Nacional de Oncología de la
Habana, en los años 1997 y 1998, con sintomatología de obstrucción nasal de
entre 8 meses y 1 año de evolución, detectándose en ambos casos lesiones del
macizo facial y cuyo diagnóstico fue: Plasmocitoma.
El primer caso se trataba de una paciente del sexo femenino,
de 48 años de edad, con una tumoración del seno maxilar derecho con destrucción
de todas sus paredes y en la que fue necesario una Maxilectomia Derecha para
concluir el diagnóstico. Durante el estudio posterior se encontró un tumor abdominal,
planificándose una Laparatomía Exploradora que teniendo en cuenta los hallazgos
transoperatorios, se realizó una Histerectomía total con doble anexectomía cuyo
resultado anatomopatológico fue: Cistadenoma Seroso; en la actualidad se encontran
completando su estudio.
El segundo caso, se trataba de un paciente del sexo masculino
de 54 años de edad, con una tumoración de la región medio facial al cual se
le planificó tratamiento combinado con poliquimioterapia y radioterapia con
Co60, encontrándose actualmente controlado de su enfermedad. En ambos pacientes
no se encontró presencia de lesión a otro nivel en examen físico y estudio radiografico,
biopsia de medula ósea normal, función renal conservada, niveles de calcio y
hemoglobina dentro de limites normales.
PALABRAS CLAVE
Plasmocitoma.
INTRODUCCIÓN
El Mieloma Múltiple ( Enfermedad de Kahler ) es un tumor del
esqueleto originado en las células reticuloplasmáticas de la médula ósea ( plasmocitoma
), que tras una fase de tumor primario pocas veces localizado en un solo hueso,
usualmente se convierte en una enfermedad sistémica. Afecta sobre todo a sujetos
viejos y evoluciona con dolores reumatoides o neuralgiformes, anemia, velocidad
de sedimentación muy acelerada e hiperprotinemia y en más de la mitad de los
casos con una proteinuria de Bences-Jones positiva.
El comienzo de la enfermedad es en general solapado, en ocasiones
una fractura insolita es el síntoma que descubre la enfermedad; otras veces
se trata de un paciente que venía sufriendo durante varios meses dolores reumatoides
o neuralgiformes, que no ceden a los analgésicos, los que hacen sospechar la
enfermedad. Esta enfermedad es mucho más frecuente en el sexo masculino, que
en el sexo femenino. Existe hiperproteinemia, las proteínas aumentadas son anormales,
se han identificado electroforeticamente tres tipos globulínicos principales
de mieloma: los alfa , beta y gamma. La proteinuria de Bences-Jones puede ser
discontinua, la misma se trata de una proteína que coagula a 42 grados, se disuelve
a 92 grados y precipita a continuación de la ebullición al enfriarse el tubo.
Pueden observarse hiperuricemia e hiperuricosuria, las manifestaciones neurologicas
pueden desarrollarse por compresión directa de la médula espinal, raíces raquideas,
nervios craneales periféricos o bien a consecuencia de la fractura patológica
de un cuerpo vertebral o un hueso largo.
El diagnóstico del plastocitoma solitario o mieloma solitario
del hueso se realiza cuando el survey óseo no muestra lesiones adicionales.
El curso de la enfermedad es relativamente indolente. La mayor parte evoluciona
a mieloma múltiple, la progresión transcurre en un período de 2 a 10 años. El
plasmocitoma aparece ocasionalmente en la región de la cabeza y el cuello (1),
el que por su forma de presentación y exámen clínico puede ser confundido con
los tumores que se presentan habitualmente en esta región; pudiendo observarse
en las regiones fosa nasal, nasofaríngeo y seno maxilar (2).
Las modalidades terapéuticas utilizadas son la radioterapia con Co 60 y la quimioterapia.
REPORTE DE CASO CLÍNICO
Caso 1.
Paciente T.M.B, historia clínica 275645, 48 años de edad, sexo
femenino, con historia de obstrucción nasal de 1 año de evolución por lo cual
es valorado por el otorrinolaringologo y posteriormente enviado a nuestro centro,
siendo evaluado el dia 15 de abril de 1997, donde se detécta al exámen físico
abombamiento del paladar duro del lado derecho. Se realizó Tomografia Axial
Computarizada del macizo facial, evidenciándose tumoración que ocupaba todo
el seno máxilar derecho, destruyendo todas sus paredes; se realizan múltiples
biopsias por aspiración que no fueron concluyentes, por lo cual se decidió tratamiento
quirúrgico:
Maxilectomía Derecha, el informe anatomopatológico B9803747 fue Plasmocitoma
plasmocitico (lesión ósea)
Estudio inmunohistoquímica positivo para cadena Lambda
Proteina de Bences Jones.:No contiene.
Examenes de laboratorio: Dentro de limites normales.
Biopsia de medula ósea: Integridad de las tres series.
Survey óseo: Acentuados cambios artrósicos.
US de Abdomen. Por detrás y a la derecha de la vejiga se observa una imagen
ecolucida de 125 por 89 por 133 milimetros , sin poderse precisar su origen.
Laparoscopía. Quiste gigante de cavidad pélvica y hemiabdómen inferior, de posible
origen ginecológico.
Se decidió Laparotomía Exploradora. - Histerectomía Total con doble anexectomía;
el 29 de enero de 1999 .
Informe A.P. (99-795,796,797) Cistadenoma Seroso
Caso 2.
Paciente A.M.G, historia clínica 283947, 54 años de edad,
sexo masculino, con historia de obstrucción nasal de 8 meses de evolución por
lo cual es valorado por el otorrinolaringó y posteriormente enviado a nuestro
centro, siendo evaluado el dia 17 de agosto de 1998, donde se detécta una lesión
que ocluye la fosa nasal derecha y se extiende a la nasofaringe. Se realiza
Tomografía Axial Computarizada que en los cortes coronales muestra una extensa
lesión en la zona medial del macizo facial, que infiltra el seno maxilar derecho
así como la base del cráneo, desplazando el globo ocular hacia fuera y adelante.
Se decidió tomar muestra del tejido de la fosa nasal el 17 de agosto de 1998.
Informe AP. no. 9807444 Plasmocitoma plasmoblastico(lesión de tejidos blandos)
Estudio inmunohistoquímica positivo para cadena Lambda
Survey óseo: No alteraciones.
Proteinas de Bences jones: No contiene.
Examenes de laboratorio: Dentro de limites normales.
Biopsia de medula ósea: Integridad de las tres series.
Se decide tratamiento combinado, poliquimioterapia con Vincristina , Alkeran,
Ciclofosfamida y Prednisona, con posterioridad al tratamiento radiante con Co60,
recibiendo tres ciclos de quimioterapía.
Actualmente se encuentra controlado de su enfermedad.
Diagnóstico histológico : Plasmocitoma.
ANATOMÍA PATOLÓGICA
Descripción macroscópica
Caso # 1
Biopsia de maxilar. 98-3747-48
3747 Rotulado Seno Maxilar.
Pieza qurúrgica de maxilarectomía que incluye segmento de paladar duro con canino
y 2 molares que mide 6 X 6 X 5 cm. que al corte muestra una tumoración blanquecina
con áreas de necrosis y hemorragia en toda su extensión. En el mismo frasco
se reciben fragmentos laminares de hueso. (11-1).
3748 Rotulado borde de sección.
Fragmento de tejido de color pardo de 1 cm. (3-1)
Caso # 2
98-7444 Rotulado fosa nasal
Fragmento de tejido de color pardo de 8 mm.(1-1)
Descripción microscópica.
Microscópicamente el tumor consiste en láminas de células plasmáticas
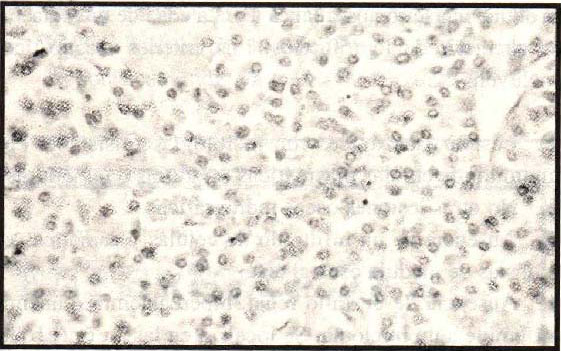
con grados variables de diferenciación, estroma vascular (Figura
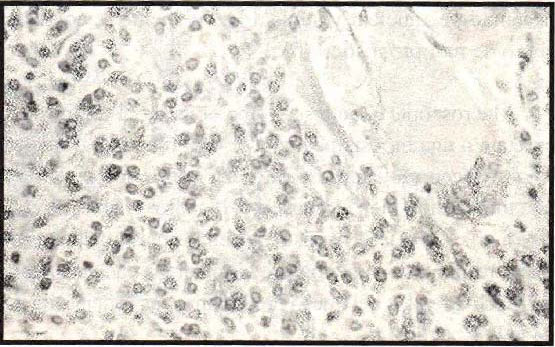
1), con un componente estromal mínimo, el amiloide está presente en un 25%
(Figura 2); las mitosis son muy raras. Las lesiones pueden
ser bien diferenciadas plasmacíticas (caso #
1) o poco diferenciadas plasmoblásticas (caso
# 2) en el primer grupo las células plasmáticas más maduras tienen núcleo
excéntrico, abundante citoplasma, halo perinuclear claro, correspondiente al
área de Golgi; otras células tienen la cromatina más dispersa e irregular, halo
ausente, forma más poligonal, pudiendose observar formas binucleadas o trinucleadas;
en el segundo grupo las células son de aspecto más inmaduro con cromatina nuclear
fina y nucleolo prominente o más gruesa agrupada e irregular (3-4).
Los estudios de las células plasmáticas por inmunohistoquímica
e inmunofluorescencia demuestran positividad para el CD 38, PCA-1, PC-1, inmunoglobinas
citoplasmaticas y la negatividad para el HLA-DR, CD 19, CD20, CD 22, CD 24,
CD 25 e inmunoglobina de superficie. Se ha detectado un patrón heterogéneo de
expresión de los antígenos de superficie, CD 33 y CD 13 (antígenos mieloides
específicos), CD 10 (antígeno presente en la etapa temprana de diferenciación
de la célula B) y CD 38 (antígenos mielomonocíticos) (5).
DISCUSIÓN
El plastocitoma solitario o mieloma solitario del hueso, es
una neoplasia de células plasmáticas que produce una lesión ósea o de tejidos
blandos única.(3)La edad de presentación es alrededor
de los 50 años más frecuente en hombres. Los criterios diagnósticos incluyen:
1)-Lesión única, con histología consistente con un tumor de
células plasmáticas (maduras o inmaduras).
2)-No otras lesiones óseas radiográficas.
3)Ausencia de un infiltrado de células plasmáticas en biopsias de médula ósea
al azar.
4)-Ausencia de un fallo renal, hipercalcemia o anemia atribuible a un mieloma.
Se localiza en las vértebras torácicas, lumbares, costillas, escápulas, huesos
pélvicos, cráneo, mandibula y huesos largos. (3,4,5)
El plasmocitoma solitario se localiza en el hueso o en las partes blandas (extramedular).
En el segundo caso son más frecuentes en las mucosas de las vías aéreas superiores
(fosa nasal, seno maxilar, nasofarínge).Puede haber extensión en el hueso adyacente.
Menos del 50 % de los pacientes tienen proteína monoclonal detectable en suero
y orina; la presencia de inmadurez nuclear y nucleolo prominente se considera
signo de mal pronóstico. (5,6).
Macroscópicamente se recibe el material en forma de curetaje
o una biopsia pequeña de tejido blando, rojo gris, rojo-grisáceo moteado. En
la pieza quirúrgica se observa una tumoración bien circunscrita, blanda o friable,
de color rojo a gris. (3) (4).
Se ha visto que en los plasmocitomas de tejidos blandos, predominan las cadenas
ligeras de la IGA y en los del hueso predominan las cadenas ligeras de la IGC.
(6).
El diagnóstico diferencial incluye (7,8,9)
1) Osteomielitis crónica.
2) Gammapatía monoclonal de origen indeterminado y la plasmocitosis reactiva.
3) Tumor metastásico. Carcinoma indiferenciado de la región de cabeza y cuello.
4) Estesioneuroblastoma olfatorio.
5) Linfoma de células grandes tipo inmunoblástico.
6) Linfoma linfoplasmocitoide (Macroglobulinemia de Waldenström).
7) Enfermedades de cadenas pesadas.
8) Granuloma de celulas plasmatica.
El diagnostico diferencial entre el granuloma de células plasmática
y el plasmocitoma plasmocitico es dificil siendo imprecindible en ocasiones
el estudio Inmunohistoquimico que demuestra una población policlonal balanciada
kappa y lambda en el primero.
EVOLUCIÓN
La evolución de ambos casos ha sido satisfactoria
CONCLUSIONES
El plasmocitoma solitario del área de cabeza y cuello, es una
entidad poco frecuente, pero que debe tenerse en cuenta en el diagnóstico diferencial
al evaluar los tumores del macizo facial.
BIBLIOGRAFÍA
- Miller F R, Lavertu P, Wanamaker J
R, Bonafede J, Wood B G: Plamacytomas of the
head and neck. Otolaryngol Head Neck Surg 1998, 119 (6): 614- 618.
- Nowak Sadzikowska J, Weiss M :
Extramedullary plasmacytoma of the larynx. Analysis of 5 cases. Eur
J Cancer 1998, 34 (9): 1468.
- Fechner R E, Mills S E : Tumors
of the bones and joints. Lesions of Hematopoietic, Lymphoid and Histiocytic
Elements. Atlas of Tumor Pathology. Third Series. Fascicle 8 Armed
Forces Institute of Pathology. Washington, D.C. 1992 : 211-232. 368.
- Brunning R
D, Mc Kenna R W.: Tumors of the bone marrow.
Plasma cell dyscrasias and related disorders. Atlas of tumor pathology.
Third series Fascicles 9. Armed Forces Institute of Pathology, Washington,
D.C., 1993 : 323 -
- Brunnig R D : Bone Marrow In: Ackerman
s Surgical Pathology. 7 th ed. Vol.2. ST Louis: C.V. Mosby, 1989: 1379
- 1453.
- Roca Estellés M J, Meseguer García
P, García Herreras F, et al: Plasmocitoma extramedular.
Poster electrónico 019. CD-Rom del II Congreso Virtual Hispanoamericano de
Anatomía Patológica (1 de junio - 31 de julio de 1998) (CIUDAD REAL, ESPAñA).
HTTP: // WWW. CONGANAT. ORG/ ¡¡ Congreso.
- Harris L N, Jaffe E S, Stein H, et
al: A revised European-American Classification
of Lymphoid Neoplasms: A preposal from the International Lymphoma Study Group.
Blood, vol 84 No.5, septiembre 1994: pp 1361-1392.
- Alvaro T, Basch R, Salvadó M T , Martínez
S: Perfil morfológico e inmunofenotípico de
los síndromes linfoproliferativos. Marcadores , morfologia, inmunohistoquímica
y diagnóstico diferencial de la enfermedad de Hodgkin, linfomas no Hodgkin
y síndromes linfoproliferativos extranodales. Tercer curso de hematopatología.
Tortasa, Ins. Cat, Salut, 1996, p. 111- 160.
- Rosas- Uribe A. et al :
La Biopsia por punción de la médula ósea en patología quirúrgica I. Normal
y no Neoplásica. Patología (Méx.) 1991,29:29-38.
FIGURAS
|

Figura 1. Tumoración constituida por
abundantes células plasmáticas,con formas binucleadas y
trinucleadas,presencia de material amicloide. Se observa la disposición
de las células neoplásicas alrededor de una estructura vascular.
|
|

Figura 2 . Abundantes células plasmáticas,
amiloide y células gigantes multinucleares alrededor de un depósito
de material amiloide.
|