OBJETIVOS
Exposición del estudio clínicopatológico de dos casos de carcinoma de células renales cromófobas con transformación sarcomatoide y discusión, a partir del análisis clínico-patológico de nuestros casos y de la revisión de la literatura, sobre si existen diferencias pronósticas con otros carcinomas sarcomatoides y en qué factores histopatológicos pueden basarse tales diferencias.
MÉTODOS
En nuestra serie revisada de 139 carcinomas de células renales extirpados quirúrgicamente durante el período 1977-1999 figuran 2 casos de carcinoma de células cromófobas con transformación sarcomatoide, lo que supone un 15% de 13 casos totales de carcinoma de células cromófobas y un 18% de 11 casos de carcinomas renales sarcomatoides en la misma serie.
RESULTADOS / CASOS CLÍNICO-PATOLÓGICOS
El primer caso corresponde a un varón de 73 años con un tumor localmente avanzado no metastásico que fue resecado de forma paliativa. Histológicamente muestra un predominio de la masa sarcomatosa, con pequeños focos epiteliales en los que se reconocen las características morfológicas, histoquímicas e inmunohistoquímicas del carcinoma de células cromófobas. El enfermo falleció tras un seguimiento de 11 meses.
En el segundo caso, correspondiente a una mujer de 70 años, el tumor debutó con dolor costo-lumbar y episodios aislados de macrohematuria. Anatomopatológi-camente se trata de un carcinoma de células cromófobas reconocible con áreas sarcomatosas focales. Su estadío era pT2pN0M0 y la paciente se encuentra libre de enfermedad a los 46 meses de seguimiento.
El estudio inmunohistoquímico de ambos casos demuestra, en el componente sarcomatoso, fuerte positividad para vimentina y, focalmente, para EMA, positividad aislada para AE1 y AE3 solo en el segundo caso y negatividad para actina, desmina y mioglobina. El componente epitelial es positivo para AE3 y EMA y negativo para AE1, vimentina y CD68.
CONCLUSIONES
La mayoría de los casos publicados de carcinoma de células cromófobas con transformación sarcomatoide son similares a nuestro primer caso, es decir, muestran masas sarcomatosas con identificación focal de carcinoma y, generalmente, mala evolución. Pensamos que, como en todo carcinoma de células renales, el pronóstico dependerá del estadío y grado tumorales (que en el caso de los carcinomas renales con transformación sarcomatoide correspondería al grado máximo) y que posiblemente poco pueda influir el tipo de componente epitelial detectado (sea éste de células cromófobas, claras, cromofílicas, etc.). Sin embargo, es imposible descartar que la proporción relativa de los componentes epitelial y sarcomatoso pueda tener algún tipo de influencia en estos tumores.
carcinoma de células renales sarcomatoide, carcinoma de células cromófobas.
El carcinoma de células cromófobas (CCCr) es un raro subtipo de carcinoma de células renales (CCR) con unas características especiales microscópicas y ultraestructurales(1),(2) y un pronóstico en general favorable, mejor al del CCR de células claras o granulares, también llamado hipernefroma "clásico". La transformación sarcomatoide se ha descrito en todas las variantes del CCR, incluyendo el hipernefroma convencional, el carcinoma cromofílico y de conductos colectores(3). La transformación sarcomatoide del CCCr se ha descrito recientemente (4) ; hay 9 publicaciones sobre el tema, siete de ellas en lengua inglesa (3-8),(12) , con un total de 16 casos(3),(9),(12) (12 en la literatura inglesa). La mayoría son carcinomas sarcomatoides en los que se identifican islotes epiteliales con las características del CCCr. En tres de ellos, al menos, se trataba sin embargo de CCCr con focos menores de transformación sarcomatoide. Todos estos casos se han estudiado desde los puntos de vista histoquímico, inmunohistoquímico y citométrico. En el presente trabajo revisamos diferentes aspectos de los casos publicados y los comparamos con dos casos propios, un CCCr con áreas sarcomatoides extensas predominantes, en estadío pT4, que falleció pocos meses después de la resección quirúrgica, y un CCCr en estadío pT2N0M0 con seguimiento de 46 meses.
De nuestra serie revisada de 139 carcinomas renales extirpados quirúrgicamente en el Hospital del Área Norte de Las Palmas durante el período comprendido entre septiembre de 1977 y octubre de 1999, 11 casos (8%) corresponden a carcinomas sarcomatoides (CS) y 13 (9,3%) a CCCr, y se identificaron 2 casos de CCCr con transformación sarcomatoide (15% de los CS, 18% de los CCCr y 1,36% del total de CCR). Se revisaron sus historias clínicas, los informes anatomopatológicos y la totalidad del material del archivo de preparaciones. Se obtuvieron nuevas secciones de los bloques de parafina que contienen tumor (8 en un caso y 9 en el otro) y se realizaron tinciones especiales (Hierro Coloidal, PAS, PAS-diastasa y Azul-Alcián) e inmunotinción para citoqueratinas de alto y bajo peso molecular (AE1, AE3, BioGenex), antígeno epitelial de membrana (EMA, Dako), vimentina (Dako) y antígeno marcador de actividad macrofágica lisosomal CD68 (clon kappa-1, Dako), tanto en secciones tomadas del componente epitelial como del sarcomatoide. En el primer caso, además, se sometió el componente sarcomatoide a inmunotinción para actina (Dako), desmina (Dako) y mioglobina (BioGenex).
CASOS CLÍNICOS.
CASO Nº 1: Varón de 73 años sin antecedentes urológicos de interés que presentaba síndrome constitucional, hematuria macroscópica y dolor lumbar derecho. En la exploración se palpaba una masa en la fosa renal derecha. No se detectaron metástasis. La TAC demostró una gran masa de 15,5 cm. de dimensión mayor que distorsionaba la silueta renal, parecía englobar la glándula suprarrenal homolateral y se adhería a estructuras vasculares y a la pared abdominal. Se efectuó una intervención quirúrgica con fines paliativos y se extirparon la masa con la grasa de la celda renal, la glándula suprarrenal y el resto del riñón. La resección, dificultosa, se llevó a cabo en varias fracciones. Se trataba de múltiples fragmentos, los mayores de los cuales, de 15x9x5 cm. y 11x6x4 cm., correspondían a una masa tumoral blanda, encefaloide, amarillenta-anaranjada, con áreas calcificadas y zonas de fibrosis. En uno de los fragmentos se reconoció el uréter rodeado por tumor y una porción de pelvis renal. En otro se identificó un polo renal con una zona irregular de infiltración cortical.
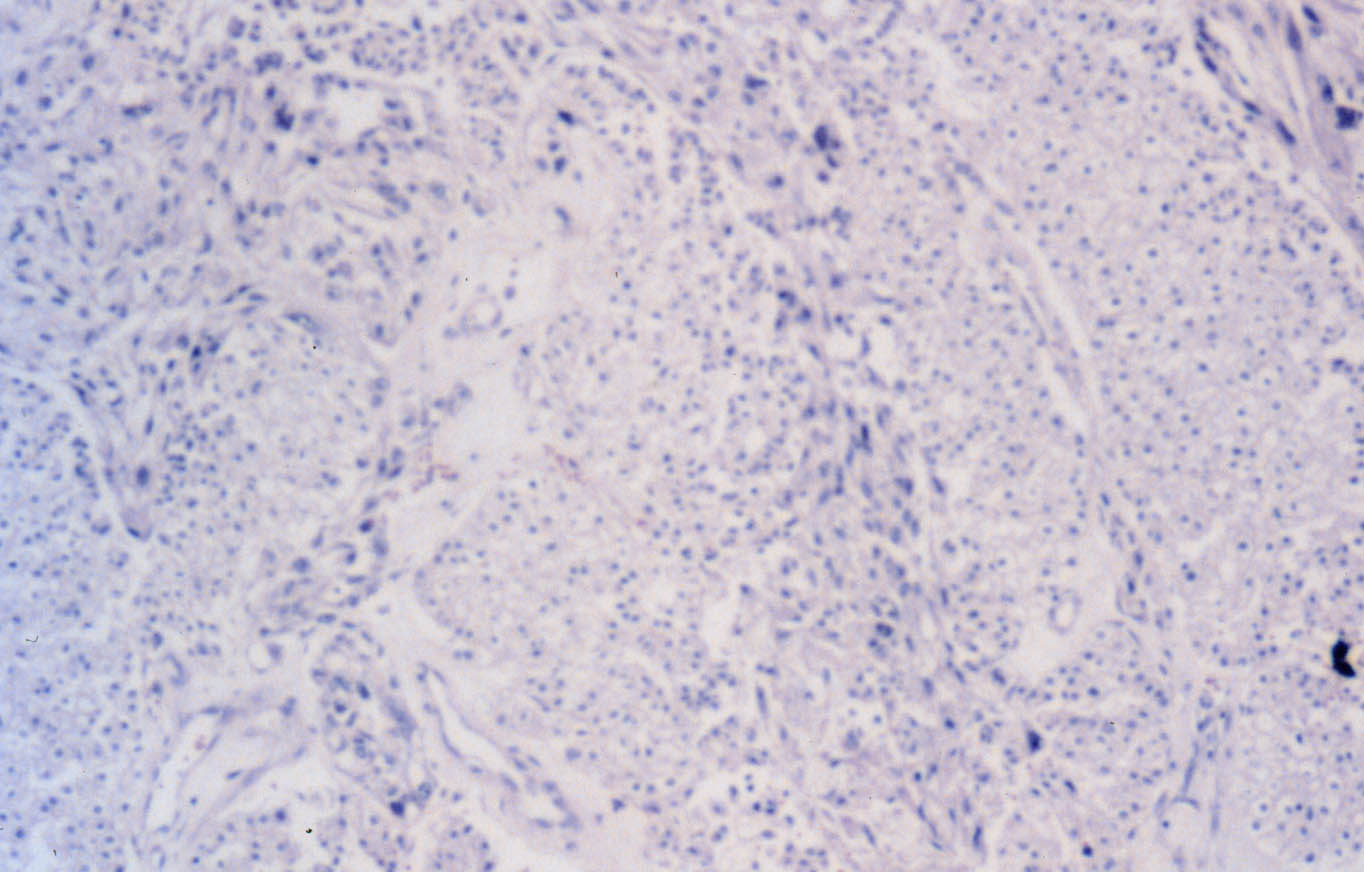
Histológicamente, en el tumor predomina una proliferación de células pequeñas, redondas o ligeramente alargadas, de escaso citoplasma acidófilo y núcleo oval con nucleolo pequeño. Las mitosis son numerosas (Figura 1). Se observan frecuentes células bi- y multinucleadas. En algunas secciones se aprecian nidos de células epiteliales de citoplasma amplio y claro, a veces con hemorragia y necrosis, que crecen en un estroma fibro-hialinizado. Estas células epiteliales muestran una delimitación neta, con membrana citoplásmica reforzada, halos perinucleares, núcleos redondeados y nucleolos poco prominentes (Figura 2). Se reconocen figuras de mitosis, pero la escasez de este componente epitelial no permite un recuento adecuado.
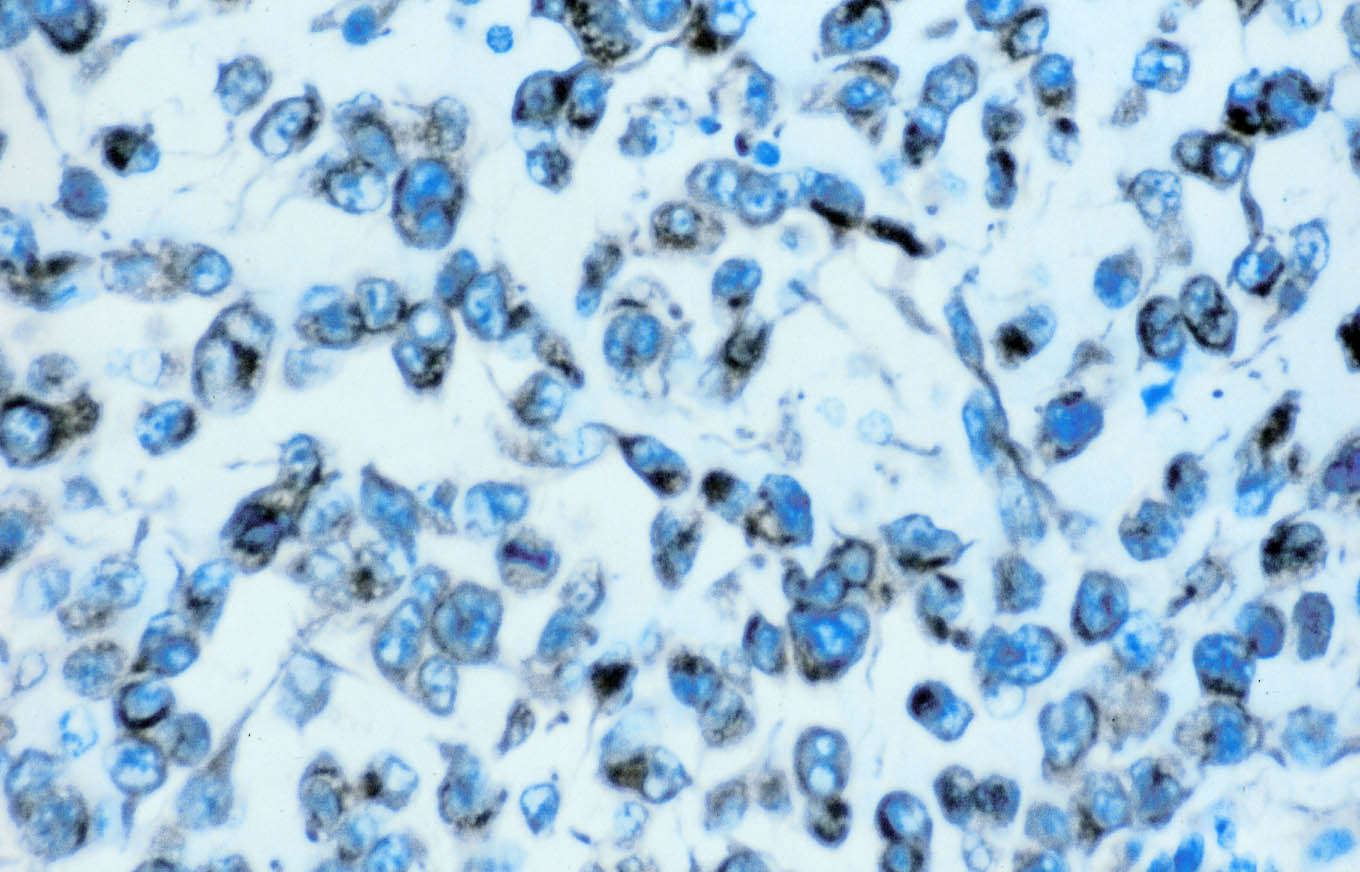
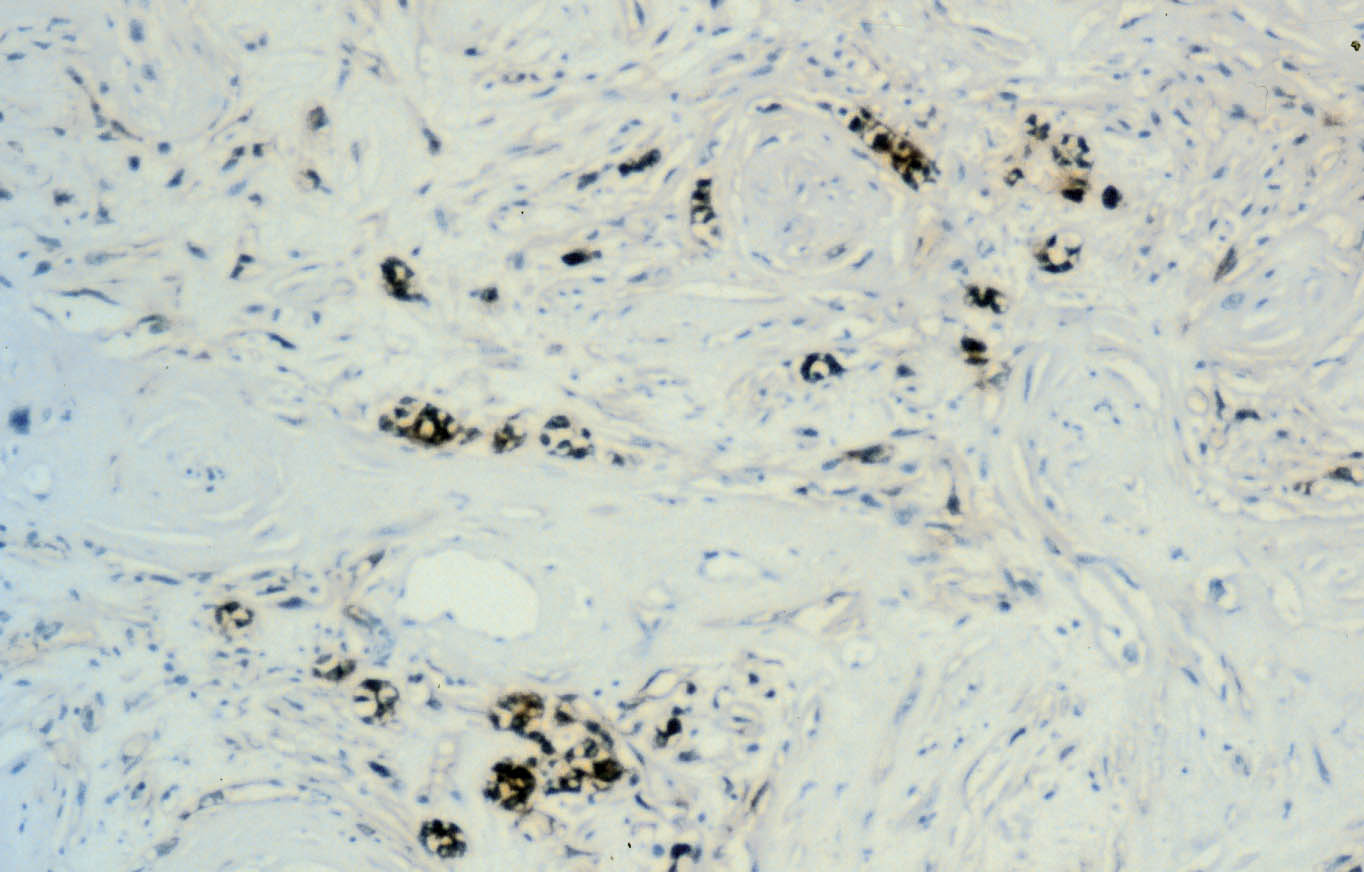
El estudio inmunohistoquímico del componente sarcomatoide revela una tinción difusa y fuerte con anticuerpo anti-vimentina, y negativa con anticuerpos antiqueratinas (Figuras 3 y 4). Se aprecia inmunotinción focal para EMA y cerca del 10% de las células muestran inmunotinción positiva para CD68, que pone de manifiesto también una marcada reacción inflamatoria en la celda renal con proliferación histiocitaria asociada. La inmunotinción es negativa para actina, desmina y mioglobina. El componente epitelial muestra inmunotinción positiva para queratinas de bajo peso molecular (AE3) y EMA, difusa y con el característico refuerzo de las membranas celulares (Figura 2), y negativa para vimentina y CD68.
El paciente falleció once meses después de la intervención quirúrgica y no se realizó estudio necrópsico.
CASO Nº 2: Mujer de 70 años con clínica de dolor costolumbar izquierdo, continuo y con exacerbaciones, de 5-6 meses que refería varios episodios de hematuria macroscópica autolimitada en el último mes. Presentaba síndrome constitucional con pérdida de 10 Kgs. en los últimos 2-3 meses. La exploración revelaba ocupación de la fosa renal izquierda. La ecografía abdominal y la TAC abdómino-pélvica pusieron de manifiesto una masa en el polo renal superior izquierdo, de 8x8x6 cm., de contornos bien definidos e interior heterogéneo, con zonas de necrosis y focos de calcificación, y adenopatías hiliares homolaterales. La gammagrafía ósea fue negativa.
En la intervención quirúrgica se practicó una nefrectomía radical izquierda con adrenalectomía y linfadenectomía de la cadena perirrenal.
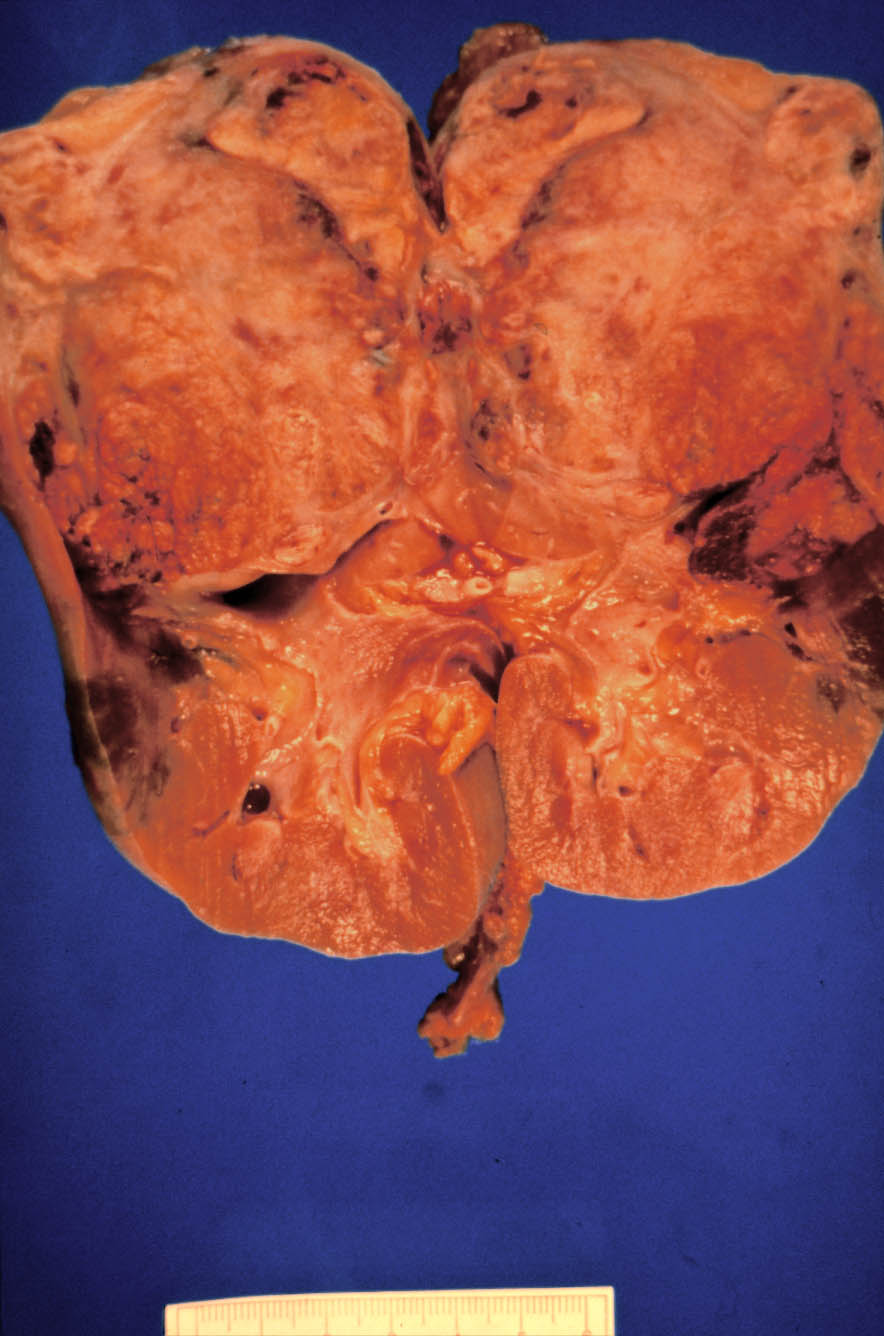
La pieza quirúrgica mostraba una masa polar superior de 8x7,5x7 cm., bien delimitada, amarillo-grisácea, con áreas de aspecto necrótico y hemorrágico (Figura 5). La glándula suprarrenal era normal. De la pieza de linfadenectomía se aislaron 15 ganglios linfáticos, no metastatizados.
Histológicamente, el tumor está formado por nidos celulares grandes separados por finos septos vasculares y constituidos por dos tipos de células: unas grandes, de citoplasma claro finamente reticulado, dispuestas preferentemente en situación paraseptal, y otras más pequeñas, de citoplasma suavemente eosinófilo, colocadas en el centro de los nidos. Todas las células de los nidos presentan aclaramiento perinuclear a modo de halo. La mayoría de los núcleos son grado 1-2 de Furhman, si bien se mezclan con ocasionales núcleos grandes e irregulares, a veces alargados y con pliegues longitudinales. No se aprecian nucleolos. La actividad mitótica es de 1,5 mitosis/10 campos de gran aumento.
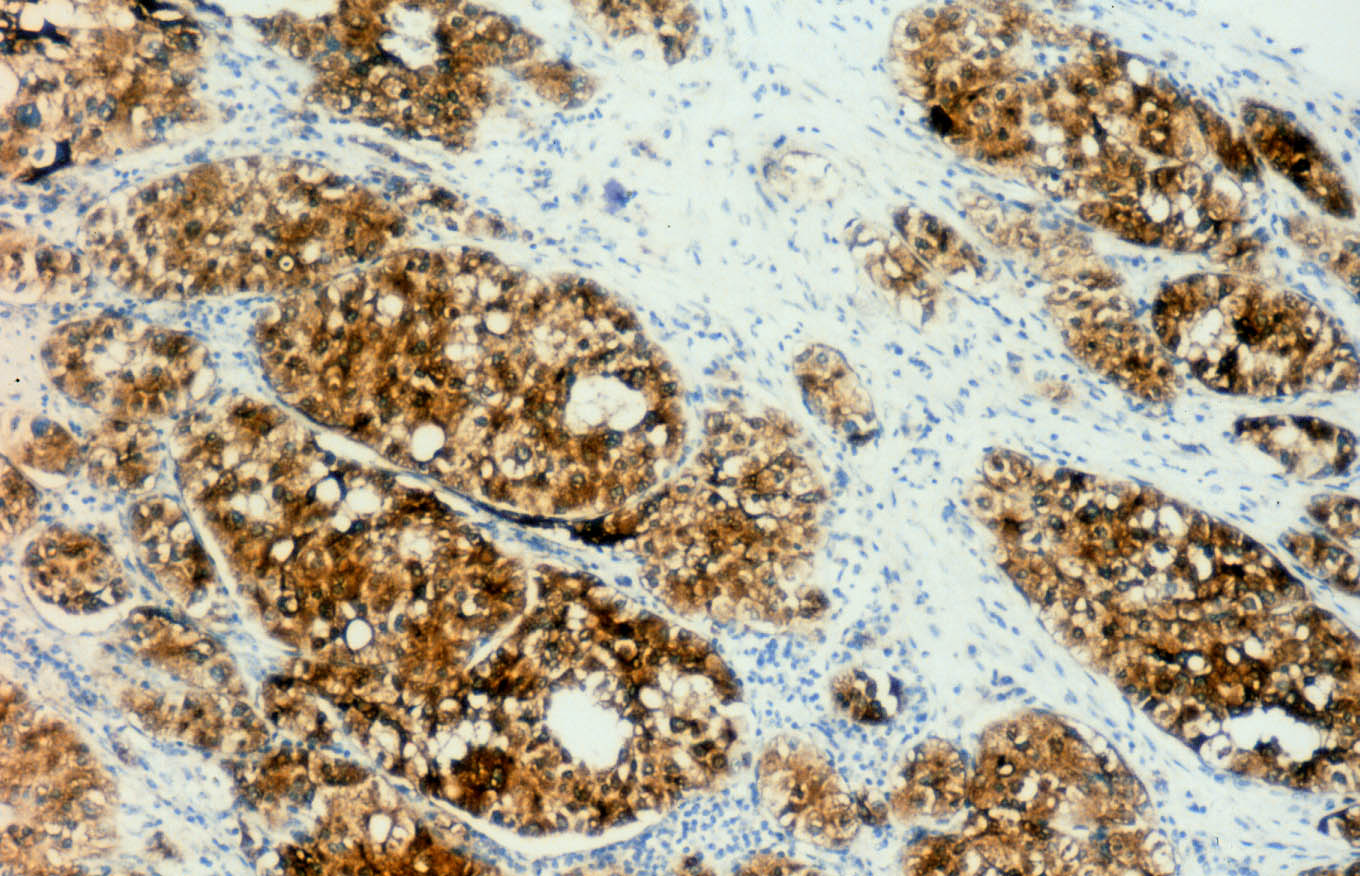
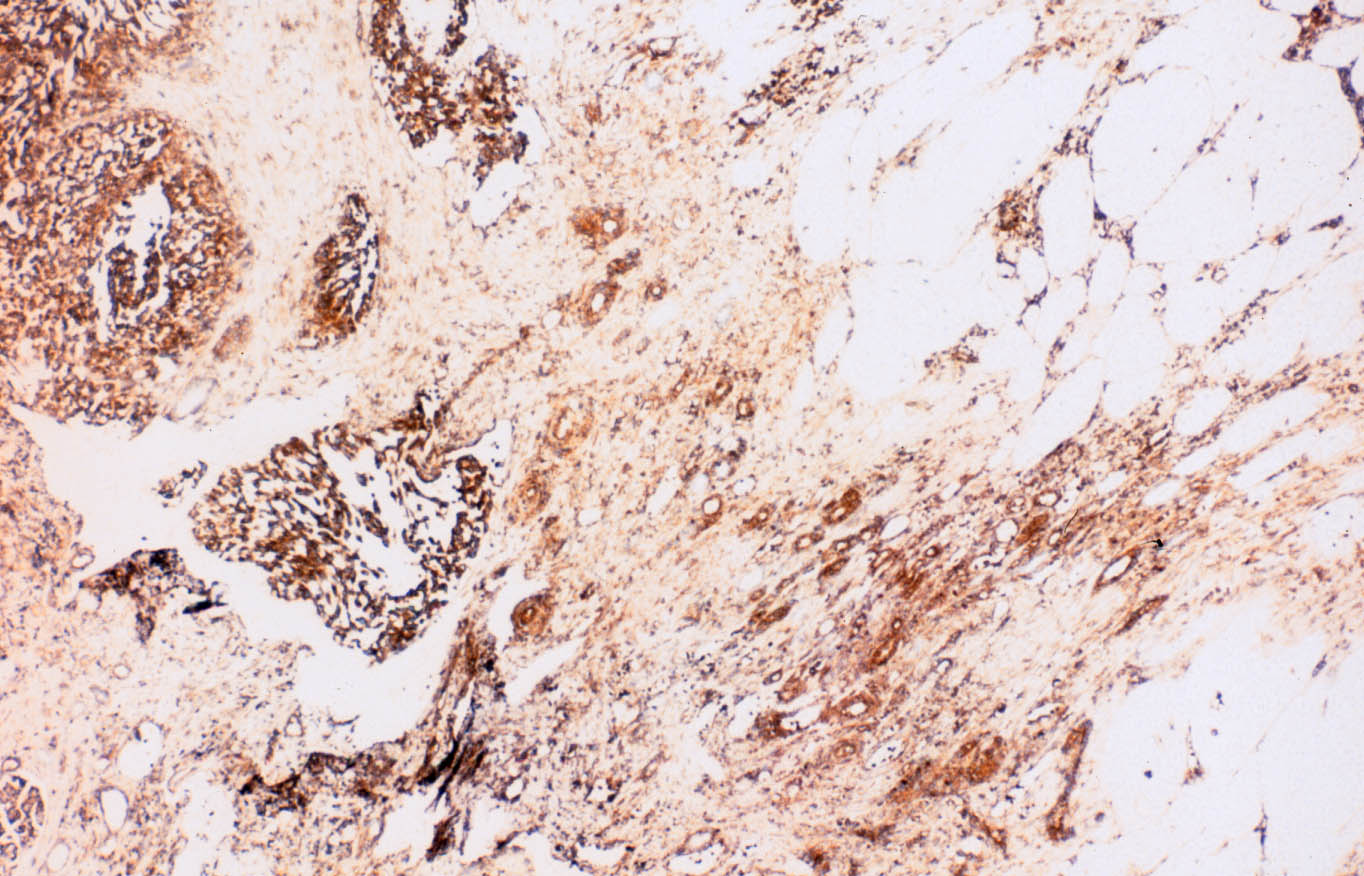
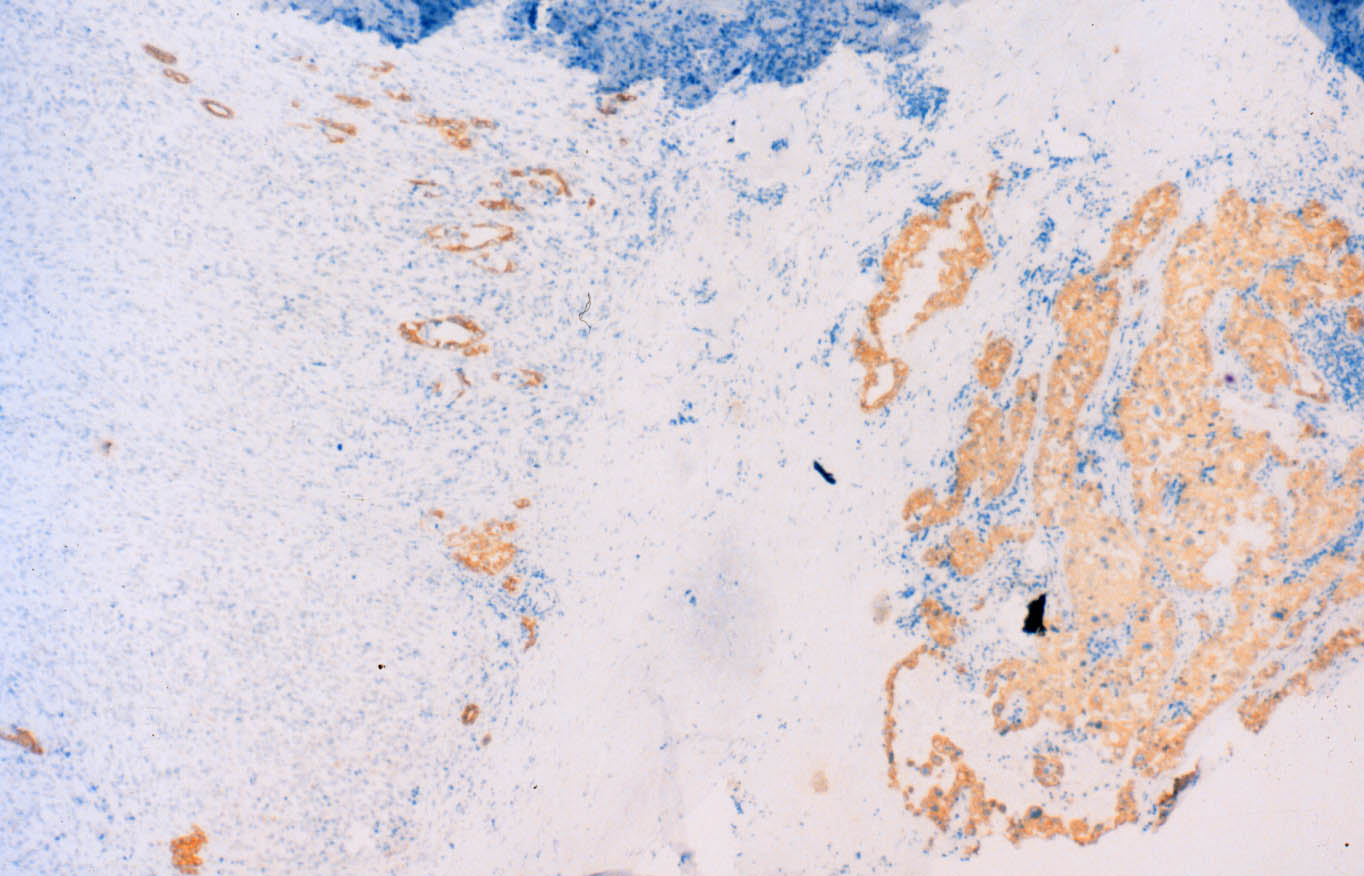
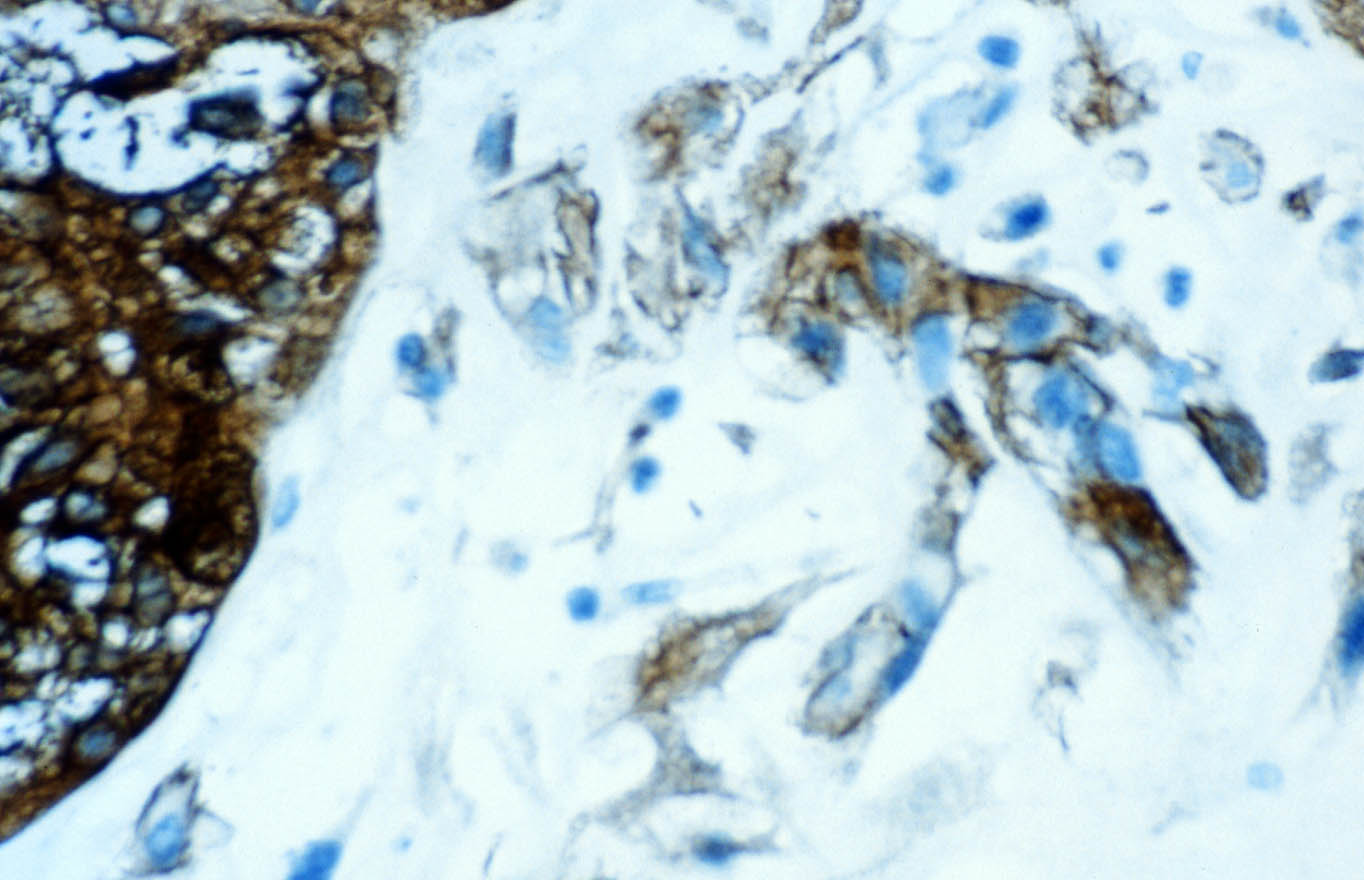
Las células se tiñen con hierro coloidal para mucinas ácidas de forma difusa e intensa. En el centro de algunos nidos se observan focos de necrosis o calcificaciones. El tumor muestra extensas áreas fibrohialinas, especialmente en la zona de protrusión en la celda renal, con depósitos cálcicos y acúmulos de macrófagos. También se aprecian áreas necróticas extensas asociadas a hemorragia. De manera focal se distingue desdiferenciación tumoral hacia células fusiformes, dispuestas en cortos fascículos (Figura 6), o hacia células redondeadas, en raqueta o pleomórficas, dispersas en una matriz hialina. Todas las células desdiferenciadas poseen núcleos grandes, irregulares, hipercromáticos, y la multinucleación y las figuras mitóticas son frecuentes. Estas áreas de aspecto sarcomatoso ocupan menos del 10% de la neoplasia. El estudio inmunohistoquímico del componente sarcomatoso revela inmunotinción intensa para vimentina (negativa en el componente epitelial) y focal o en células aisladas para citoqueratinas (AE1 y AE3) (Figura 7) y EMA (Figura 8). La inmunotinción del componente epitelial muestra fuerte positividad granular para citoqueratinas de bajo peso molecular (AE3) y EMA, con refuerzo de membrana característico (Figura 8), y negatividad para citoqueratinas de alto peso molecular (AE1) y CD68, que marca macrófagos estromales en ambos componentes tumorales y aisladas células sarcomatoides.
El CCCr se ha considerado como un carcinoma de mejor pronóstico que el resto de los CCR. El CCCr se suele asociar, en el momento del diagnóstico, a grado y estadío bajos(1),(2). No obstante, en casi todas las series se mencionan algunos casos de más alto grado y estadío. Lo mismo ocurre con los rasgos histológicos que indican agresividad. Así, aunque habitualmente el CCCr no presenta necrosis ni hemorragia importantes, ni núcleos de alto grado (según el sistema de gradación de Furhman), ni actividad mitótica relevante o atípica, ni extensión extrarrenal, ni metástasis ganglionares o a distancia, ni nódulos satélites, en varias series con un cierto número de tumores se incluye algún CCCr con alguna o algunas de estas características(10). Sin embargo se sigue insistiendo en el mejor pronóstico del CCCr, sin que hasta la fecha existan estudios prospectivos, con seguimientos prolongados, comparando grado por grado y estadío por estadío el comportamiento del CCCr respecto al hipernefroma clásico, del que se separó como entidad en 19851. Pensamos que mientras estos estudios no se realicen, tal vez no sea posible más que conjeturar sobre el comportamiento real del CCCr, habida cuenta de que muchas series provienen del estudio retrospectivo de material de archivo anatomopatológico. Nos tememos que en este tipo de estudio quizá se haya tendido a rescatar aquellos casos más similares a los descritos inicialmente por Thoenes y cols.(1),(2), es decir, de más baja agresividad histológica, rechazándose en el curso de las revisiones los casos de aspecto más agresivo (con más necrosis o hemorragia, núcleos grados 3 ó 4, mitosis frecuentes, etc.) por no amoldarse a la descripción original. Hace unos años, una publicación resaltaba la existencia de variantes "agresivas" de CCCr10: 7 casos de un total de 20 tumores con un seguimiento promedio de 60 meses habían desarrollado metástasis. Dos de los casos se asociaban a carcinoma papilar renal en el mismo riñón, ignorándose cuál de los dos tumores había metastatizado. La serie completa, de 25 tumores, incluía 9 con grado 3 de Furhman, 3 con necrosis amplia y 6 con extensión extrarrenal (pT3 del sistema de estadiaje AJCC-TNM). No se apreció correlación, sin embargo, entre estos hallazgos y el comportamiento metastásico del tumor, así como tampoco con los estudios de ploidía del DNA tumoral (10).
En principio, la transformación sarcomatoide implica un aumento en la agresividad de cualquier carcinoma de células renales, que pasaría a tener un grado máximo (grado 4/4) y muy mal pronóstico (3). Esta transformación se ha descrito en todos los tipos de CCR3, incluyendo recientemente al CCCr (3),(4). Una reciente publicación (Akhtar y cols.) (3) registra un porcentaje inusual, 60%, de carcinomas renales sarcomatoides cuyo componente epitelial es un CCCr, lo que lleva a estos autores a hablar de lo que denominan la "conexión cromófoba", para resaltar una hipotética mayor tendencia de este tipo de CCR a la transformación sarcomatoide.
En la literatura a nuestro alcance hemos encontrado 16 casos de CCCr con transformación sarcomatoide, y 12 si nos ceñimos a la literatura de habla inglesa (Tabla I). Dado lo reciente de la descripción del primer caso (4), no es de extrañar que solo 9 publicaciones (7 de habla inglesa) traten del tema (3-9),(11),(12). Esta transformación suele asociarse a un curso agresivo de la enfermedad (7),(9-11), aunque hay excepciones (5). En la mayor parte de los casos publicados se trata de neoplasias ampliamente sarcomatoides con pequeños islotes en su seno en los que se reconocen las células típicas del CCCr, con sus características reacciones histoquímicas e inmunohistoquímicas(3),(7), como en nuestro caso nº 1. En unos pocos casos, como ocurre en el nuestro nº 2, se trata de CCCr con pequeños focos sarcomatosos (8) que constituyen un 10% o menos de la masa tumoral (10),(12). En cualquier caso, lo habitual es que sean tumores grandes, mayores de 7 cm(3),(5),(7),(12). Algunos se extienden más allá de la cápsula rena (l3),(12). En algunos CCCr con transformación sarcomatoide se describen áreas focales, dentro del componente epitelial, con marcado pleomorfismo nuclear(3). En algunos casos se describen también áreas de transición entre los dos componentes, sarcomatoso y epitelial (12), aunque en general la colisión entre los dos componentes se define como abrupta(3).
El componente sarcomatoso suele ser fusocelular (3) y se ha descrito un caso con diferenciación rabdomioblástica (11). Con la técnica de hierro coloidal se observa tinción difusa e intensa en el componente epitelial y más débil y focal en el componente sarcomatoso de algunos tumores(3),(12). Los anticuerpos anticitoqueratinas de bajo peso molecular y anti-antígeno epitelial de membrana siempre ofrecen inmunotinción difusa e intensa del componente epitelia (l3),(9). En el componente sarcomatoso se describe inmunotinción débil o focal para citoqueratinas (3),(12), aunque a veces puede ser más intensa (9), y resultados variables para EMA: negativa (9) o focalmente positiva (3),(12) en algunos trabajos y francamente positiva en otros (5). La inmunotinción para vimentina es siempre negativa en el componente epitelial y positiva en el sarcomatoso(3),(5),(9),(12). Según Akhtar, ambos componentes son negativos para desmina, actina muscular específica y proteína S-1003. Todos estos hallazgos están acordes con los de nuestros casos
Solo un trabajo (3) refiere los resultados del estudio por citometría de flujo de 5 CCCr con transformación sarcomatoide: detección de líneas hipodiploides (y también clones hiperdiploides en dos casos) en el componente carcinomatoso y de picos hiperdiploides e hipertetraploides en el sarcomatoso.
Finalmente, en la literatura existe cierto debate sobre el pronóstico de los CCCr con transformación sarcomatoide. Aunque en general se admite que el pronóstico debería ser el del componente sarcomatoide (7), algunos autores han apuntado que el componente epitelial podría afectar a la conducta biológica del tumor, de modo que el pronóstico fuera mejor que el de los demás carcinomas sarcomatoides renales(5). Esta posibilidad se plantea en la reseña de un único caso (5). Pensamos que es necesario evaluar series más amplias, con seguimiento adecuado, para demostrar posibles diferencias de conducta con otros carcinomas sarcomatoides renales. Suele haber acuerdo en que el pronóstico de los CCR depende de las áreas de más alto grado histológico, sea cual sea su prevalencia (13) y a los carcinomas renales sarcomatoides se les asigna el mayor grado, pero es cierto que el componente epitelial suele ser de alto grado también (13). Esto no ocurre necesariamente en el CCCr con transformación sarcomatoide, probablemente por el hecho de que de otro modo no sería reconocible como tal CCCr. Sin embargo, las proporciones relativas de los dos componentes son muy variables, lo mismo en los CCCr con transformación sarcomatoide que en los CCR de otro tipo con idéntica transformación. Quedaría por demostrar si el porcentaje del componente sarcomatoso o su volumen en términos absolutos puede influir en el CCCr con transformación sarcomatoide. Como se refleja en la diferente evolución de los dos casos aquí presentados, entra dentro de lo posible que no quepa esperar la misma evolución en tumores ampliamente sarcomatosos con un componente epitelial mínimo que en CCCr con transformación sarcomatosa microscópica. Es decir, que quizá en el caso del CCCr con transformación sarcomatoide podría influir más en el pronóstico la cantidad respectiva de los dos componentes neoplásicos (de células cromófobas y sarcomatoide) que el hecho de que el componente epitelial originario corresponda a un CCCr. Esto deberá ser comprobado en estudios posteriores.
1.- Thoenes W, Störkel S, Rumpelt HJ: "Human chromophobe renal cell carcinoma". Virchows Arch B (Cell Pathol), 48: 207-17, 1985.
2.- Thoenes W, Störkel ST, Rumpelt HJ y cols.: "Chromophobe cell renal carcinoma and its variants. A report of 32 cases." J Pathol, 155: 277-87, 1988.
3.- Akhtar M, Tulbah A, Kardar AH y cols.: "Sarcomatoid renal cell carcinoma: The chromophobe connection". Am J Surg Pathol, 21: 1188-95, 1997. (**)
4.- Akhtar M, Kfoury H, Kardar A y cols.: Sarcomatoid chromophobe cell carcinoma of the kidney". J Urol Pathol, 4: 155-66, 1996.
5.- Gómez-Román JJ, Mayorga-Fernández M, Fernández F y cols.: "Sarcomatoid chromophobe cell carcinoma: immunohistochemical and lectin study in one case". Gen Diagn Pathol, 143: 63-9, 1997.
6.- Aizawa S, chigusa M, Ohno Y y cols.: "Chromophobe renal cell carcinoma with sarcomatoid component. A report of two cases." J Urol Pathol , 6: 51-9, 1997.
7.- Hirokawa M, Shimizu M Sakurai T y cols.: "Sarcomatoid renal cell carcinoma with chromophobe cell foci. Report of a case". APMIS, 106: 993-6, 1998.
8.- Kuroda N, Hayashi Y, Itoh H: "A case of chromophobe renal cell carcinoma with sarcomatoid foci and a small daughter lesion". Pathol Int, 48: 812-7, 1998.
9.- Joubert M, Cassgnau E, Boullanger P y cols.: "Sarcomatoid variant of chromophobe renal cell carcinoma. Report of 2 cases". Ann Pathol, 17: 392-5, 1997.
10.-Renshaw AA, Henske EP, Loughlin KR y cols.: "Agressive variants of chromophobe renal cell carcinoma" Cancer, 78: 1756-61, 1996. (*)
11.-Hes O, Michal M, Kinkor Z y cols.: "Sarcomatous chromophobe renal cell carcinoma. Two case reports". Cesk Patol, 35: 15-9, 1999. (*)
12.-Mai KT, Veinot JP, CollinsJP: "Sarcomatous transformation of chromophobe renal cell carcinoma. (Letter)". Histopathology, 34: 557-9, 1999.
13.-Murphy WM, Beckwith JB, Farrow GM: "Tumors of the kidney, bladder and related urinary structures", fascicle 11, 3ª serie, p.124. AFIP, Washington, 1994.
CASOS PUBLICADOS HASTA 1999 DE CCCr CON TRANSFORMACIÓN SARCOMATOIDE.
|
AUTORES
|
AÑO
|
PAIS
|
Nº DE CASOS
|
|
Akhtar M y cols
|
1196
|
Arabia Saudí |
1
|
|
Aizawa S y cols.
|
1197
|
Japón
|
2
|
|
Akhtar M y cols.
|
1197
|
Arabia Saudí |
5
|
|
Joubert M y cols.
|
1197
|
Francia
|
2
|
|
Gómez Román y cols
|
1197
|
España
|
1
|
|
Hirokawa M y cols.
|
1198
|
Japón
|
1
|
|
Kuroda N y cols.
|
1198
|
Japón
|
1
|
|
Hes O y cols.
|
1199
|
Rep.Checa
|
2
|
|
Mai KT y cols.
|
1199
|
Canadá
|
1
|
TOTAL...............16 casos