Introducción. Son raros los tumores con una localización intraventricular
supratentorial primaria. Resulta de mayor frecuencia la extensión intraventricular,
entre los tumores que se comportan de este modo está el astrocitoma del hipotálamo
y del nervio óptico, linfomas, subependimomas, craneofaringiomas y carcinomas
metastásicos.
Pacientes y métodos. Se realizó un estudio retrospectivo con
las historias clínicas de 11 pacientes ingresados en el CIREN en el periodo
comprendido de Enero de 1996 a Noviembre de 1999 con diagnóstico clínico de
tumor intraventricular supratentorial. De los 11 enfermos, 8 correspondieron
al sexo masculino y 3 al femenino. La edad osciló entre 15 meses y 51 años.
La afectación ventricular fue la siguiente: cinco tumores afectaron el ventrículo
lateral y seis al tercer ventrículo. Los procederes diagnóstico terapéutico
de los casos estudiados, resecados total o parcialmente fueron los siguientes,
cinco abordajes transcalloso, cuatro abordajes transcortical, una resección
neuroendoscópica y una biopsia estereotáxica. Una vez resecado el tejido se
procesó histológicamente, a 4 de ellos se les realizó técnicas inmunohistoquímicas
y a 3 estudio por microscopía electrónica, para confirmar el diagnóstico.
Resultados. El análisis histológico fue el siguiente: 3 pacientes
con diagnóstico de neurocitoma central, 3 con quiste coloide, 1 astrocitoma
pilocítico, 1 tumor neuroectodérmico primitivo, 1 ependimoma, 1 meningioma (transicional)
y una lesión inflamatoria crónica de causa no precisada. Es objetivo del presente
trabajo comunicar nuestra experiencia, así como confirmar que las lesiones localizadas
en los ventrículos laterales y en el tercero son raras si tenemos en cuenta
que se trataron 109 lesiones tumorales del Sistema Nervioso en nuestro centro
en el tiempo referido anteriormente.
Palabras Claves: Neuropatología, Tumor cerebral, Tercer ventrículo, Ventrículos laterales.
Son raros los tumores con una localización intraventricular supratentorial primaria (1). Resulta de mayor frecuencia la extensión intraventricular, entre los tumores que se comportan de este modo podemos mencionar al astrocitoma del hipotálamo y del nervio óptico, linfomas, subependimomas, craneofaringiomas y carcinomas metastásicos (2).
Selección de los pacientes:
Se realizó un estudio retrospectivo con las historias clínicas de 11 pacientes ingresados en el CIREN en el periodo comprendido de Enero de 1996 a Noviembre de 1999 con diagnóstico clínico de tumor intraventricular supratentorial. De los 11 enfermos, 8 correspondieron al sexo masculino y 3 al femenino. La edad osciló entre 15 meses y 51 años.
Tabla 1. Resumen Clínico.
Técnica quirúrgica:
Los procederes diagnóstico terapéutico de los casos estudiados, resecados total o parcialmente fueron los siguientes, cinco abordajes transcalloso, cuatro abordajes transcortical, una resección neuroendoscópica y una biopsia estereotáxica.
Procedimiento histológico:
La tabla muestra el seguimiento realizado con el tejido resecado en los 11 pacientes dadas las diferencias en el procesamiento. Todos fueron fijados en formaldehído al 10 %, embebidos en parafina, cortados en un micrótomo entre 6 y 8 micras y teñidos con Hematoxilina/Eosina. Las técnicas auxiliares sirvieron para confirmar la sospecha diagnóstica o para realizar este.
Tabla 2. Procesamiento histológico
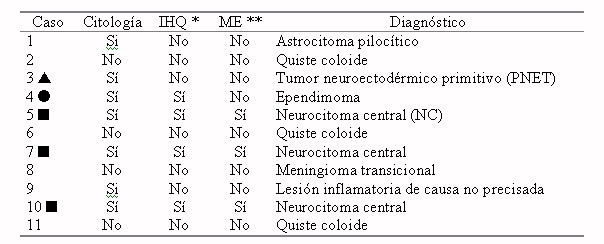

Los casos 3, 5, 7, 8, 10 presentaban signos y síntomas de hipertensión endocraneana. A los casos 1, 3, 4, 5, 7, 9 y 10 se les realizó extendido citológico transoperatorio siendo muy celular excepto para el caso 9 que fue ligera, con particularidades según el diagnóstico:
IHQ:
ME:
Dada la variedad en el diagnóstico histológico resulta conveniente particularizar algunos aspectos brevemente en cada uno de ellos.
El astrocitoma pilocítico se localiza a través del neuroaxis. Al microscopio óptico encontramos dos variantes, la juvenil y la tipo adulto. La primera afecta a niños y a adultos jóvenes, al óptico encontramos astrocitos estrellados en áreas microquísticas que alterna con zonas sólidas de células elongadas donde encontramos las fibras de Rosenthal y los cuerpos granulares eosinofílicos. En los tumores de larga evolución podemos ver pleomorfismo nuclear. La variante tipo adulto es propensa a la degeneración maligna y no existe acuerdo en cuanto a si representa una entidad clinicopatológica definida, esta forma maligna es rara. Debe ser diferenciado con el astrocitoma fibrilar y el xantoastrocitoma pleomórfico (3).
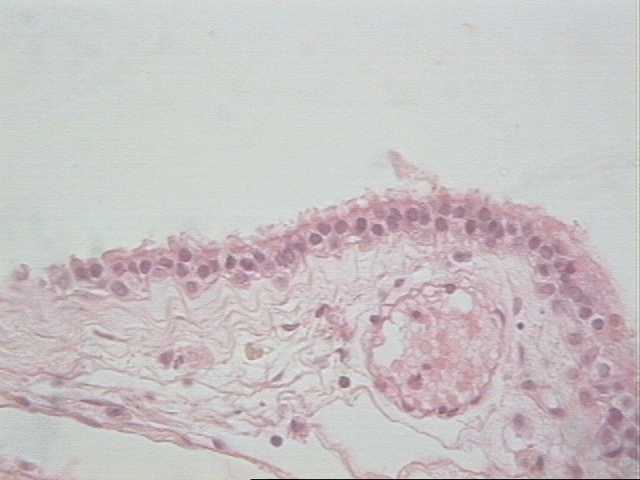
El quiste coloide, referido también como quiste neuroepitelial se localiza preferentemente en la parte anterior del tercer ventrículo aunque lo podemos encontrar en cualquier parte del sistema ventricular. Es asintomático, si crece los suficiente y obstruye el agujero del Monro puede provocar una hidrocefalia aguda. Se presenta entre la tercera y cuarta década de la vida. Le corresponde el 0.5 % de todos los tumores intracraneales. Macroscópicamente se presenta como una lesión esférica, su tamaño oscila entre 0.3 a 3-4 cm, el contenido del quiste es un líquido claro o lechoso. Histológicamente la pared del quiste es delgada, formada por un tejido fibroso, el epitelio falta en algunas zonas y varía de cúbico bajo a columnar alto, en ocasiones es ciliado, en los tres casos estudiados por nosotros estuvieron presentes los cilios (Fig. 5). La resección quirúrgica total es curativa en aquellos casos donde no existe daño en áreas aledañas a la lesión (4, 5).
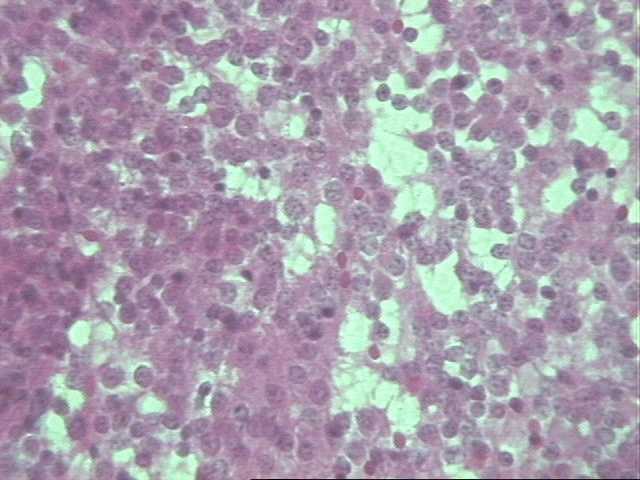
El tumor neuroectodérmico primitivo agrupa a una serie de tumores que se presentan durante el desarrollo embrionario, fetal o postnatal temprano, por lo que la mayoría de los casos se presentan por debajo de los 10 años, se han reportado 8 casos que superan los 15 años uno de los cuales tenía 57 años. Se reconocen 5 categorías clinicopatológicas: meduloepitelioma, meduloblastoma, neuroblastoma, espongioblastoma polar y el ependimoblastoma, siendo imposible en ocasiones reconocer alguna de estas variantes a pesar de realizar técnicas IHQ (diferenciación glial y neuronal) y de ME. El caso visto en nuestro hospital no pudo estudiarse adecuadamente por artefacto de electrocoagulación presente en la muestra de parafina por lo que no pudimos excluir un neuroblastoma. Todos se caracterizan por un rápido crecimiento y diseminación a través del líquido cefalorraquídeo y metástasis extracraneales en algunos casos. Al microscopio óptico encontramos un tumor muy celular compuesto por células redondas (derivan de precursoras neuroectodérmicas), pequeñas con escasa cantidad de citoplasma y núcleo redondo u oval y mitosis abundantes. El pronóstico en general es malo, no se tiene suficiente claridad de este en relación a cada una de las variantes y al grado de diferenciación. Se estima que el tiempo de sobrevida una vez hecho el diagnóstico es de aproximadamente 8 meses, nuestro paciente tiene 12 meses de evolución (4, 6).
El ependimoma por su parte es un tumor de origen glial, de crecimiento lento, le corresponde aproximadamente el 5 % de todos los gliomas intracraneales en el adulto y entre el 8 y 9 % en niños y adolescentes. Su localización es variada, comúnmente se origina en los ventrículos (particularmente en el cuarto - niños), lo podemos ver también en la región lumbosacra de la médula espinal y a nivel del filum terminal (fundamentalmente adultos). Macroscópicamente es un tumor delimitado de consistencia blanda, friable. Histológicamente el rasgo característico es la roseta ependimaria, la cual no siempre está presente en todos los casos, como sucedió en nuestro paciente, lo más común es la disposición de las células tumorales alrededor de los vasos sanguíneos (pseudorosetas perivasculares). La variante maligna es rara y se diagnóstica cuando apreciamos invasión, una extensa necrosis y atipia citológica. La benignidad histológica no guarda relación con un comportamiento clínico benigno. El diagnóstico diferencial debe hacerse con un astrocitoma mixto (fondo fibrilar y ausencia de elementos epiteliales), neurocitoma central y el PNET (4, 7).
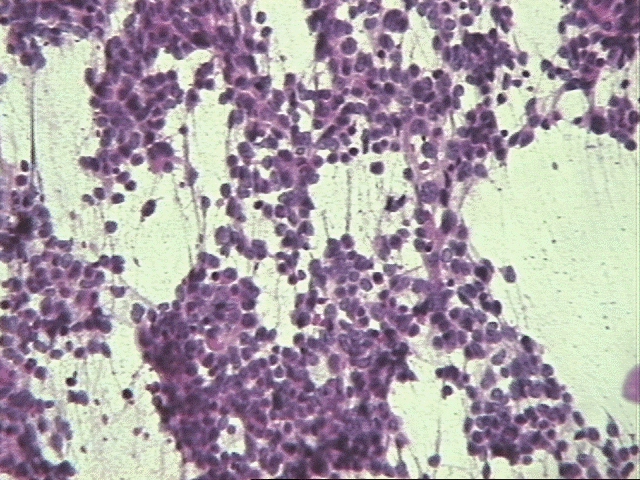
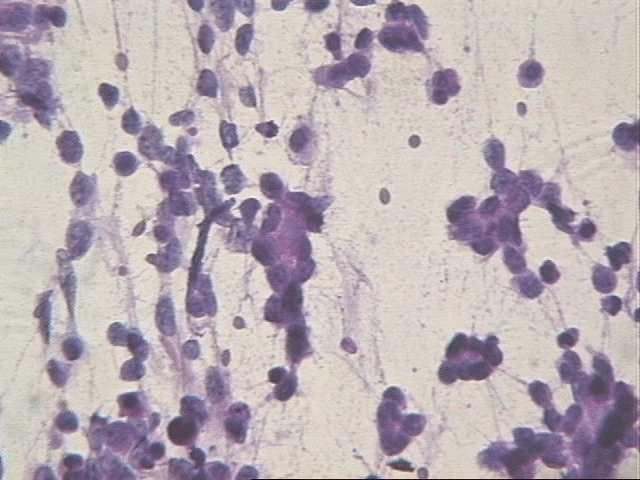
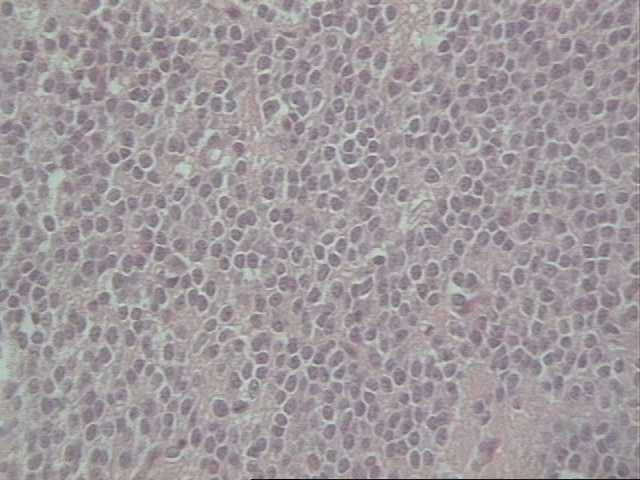
El neurocitoma central es un tumor raro, descrito por vez primera en 1982 por Hassoun y colaboradores, siendo un tumor de origen neuronal, bien diferenciado, de localización intraventricular preferentemente afectando a adultos jóvenes. El Instituto de Patología de las Fuerzas Armadas define al NC "como un tumor intraventricular discreto compuesto por células pequeñas mitóticamente inactivas. Alternativamente se le ha nombrado como neuroblastoma central y neuroblastoma bien diferenciado. Se cree que el origen de este tumor está en los remanentes neuroblásticos en la capa subependimaria o en el núcleo del septum pelúcido. El tumor tiene un comportamiento benigno con un buen pronóstico y constituye menos del 1 % de todos los tumores del Sistema Nervioso Central (SNC). Microscópicamente presenta un patrón de crecimiento sólido y monótono, de células redondas, uniformes con citoplasma granular bien definido y halos perinucleares, tendencia a la formación de estructuras rosetoides y malla fibrilar, compartiendo todos un soporte vascular florido dado por numerosos vasos capilares interpuestos. El diagnóstico diferencial del NC hay que realizarlo con las siguientes entidades: el oligodendroglioma por la presencia de calcificaciones, los halos perinucleares y la uniformidad celular; el ependimoma por su localización y las pseudorosetas y por último con el neuroblastoma con la ausencia de calcificaciones y presencia de atipia citológica, actividad mitótica, necrosis e invasión del parénquima con una afectación más extensa del sistema ventricular y al ME la ausencia de sinapsis (8, 9, 10).
La mayoría de los meningiomas ocurren en el adulto con una clara predisposición por la mujer, su localización es variada (región parasagital, convexidad cerebral, hoz del cerebro, la base del cráneo, surco olfatorio, ángulo pontocerebeloso y en el borde petroso del hueso temporal. La afectación intraventricular es rara alcanzando un gran tamaño antes de provocar los síntomas como sucedió en el caso 8. Al examen macroscópico se presenta como un tumor bien delimitado, sólido, de consistencia firme, lobulado, de color blanco grisáceo, la superficie de corte tiene un aspecto arremolinado. Al microscopio óptico en general encontramos células redondas, ovales, poligonales o fusiformes con un citoplasma pálido cuyos bordes no son precisos y un núcleo redondo u oval. Se han descrito diferentes variantes histológicas: meningotelial, transicional, fibrosa, psammomatosa, secretora, microquística, linfoplasmocitaria, cordoide, metaplásica, papilar, de células claras, con patrón pseudoglandular, esclerótica así como la maligna (frecuencia del 10 %). Diferenciarlo de un carcinoma (variante anaplásica), schwannoma (si localización intraespinal e infratentorial), ependimoma (si vemos células dispuestas alrededor de los vasos sanguíneos (11, 12, 13).
Cuando un patólogo se enfrenta al diagnóstico de un tumor cerebral debe tener en cuenta tres elementos, estos son: la edad del paciente, la localización exacta de este y el aspecto macroscópico en el momento de la cirugía (4). En cuanto a la localización, Burger y Scheithauer en el acápite de anexos resumieron de forma práctica la relación de diversos tumores que tienen una localización ventricular, permitiendo que el error diagnóstico sea menor dada la rareza de estos en comparación con aquellos tumores que tienen una ubicación hemisférica. La ubicación de estos tumores es la siguiente: ependimoma (ventrículos laterales, tercero y cuarto); subependimoma (ventrículos laterales y cuarto); astrocitoma subependimario de células gigantes - esclerosis tuberosa (ventrículos laterales); tumor del plexo coroides (niños: ventrículos laterales y tercero; adultos: cuarto); astrocitoma pilocítico (ventrículos laterales, tercero y cuarto); neurocitoma central (ventrículos laterales, raramente el tercero); tumor de células germinales y parénquima pineal (tercero posterior); craneofaringioma papilar (tercero); quiste coloide (tercer ventrículo) y meningioma (infrecuente en los ventrículos laterales, tercero y cuarto) (14).
Con este trabajo hemos confirmado que los tumores intraventriculares supratentoriales primarios son raros, nos basamos en la literatura revisada (5 años) donde existen pocos trabajos reportados con los diagnósticos vistos aquí y en nuestra propia experiencia donde tratamos 109 lesiones tumorales del SNC encontrando solamente 11 casos con esta localización en el tiempo referido.